
Original artstract by M. Shercliffe.
We’ve all experienced moments of intense stress that seem burned into our memories, whether it’s a near-miss car accident, a traumatic event, or even the anxiety of a final exam. But how does the brain cement these stressful experiences into long-term memory? The paper “Making Memories of Stressful Events: A Journey Along Epigenetic, Gene Transcription, and Signaling Pathways” [1] explores the molecular events behind this process, revealing a complex interaction between hormones, neurotransmitters, and epigenetic modifications.
Understanding the relationship between stress and memory formation begins with the hippocampus, a brain region critical for memory formation. When faced with psychological stress (like a rat being forced to swim in a lab tank, or a human speaking in front of a large crowd), the hippocampus becomes activated, in turn activating a cascade of molecular events that help encode the experience. Neurons firing in the hippocampus are not the only factor, glucocorticoids (stress hormones like cortisol in humans, or corticosterone in rats) play a significant part.
The paper highlights that these hormones don’t just passively float around, they actively shape how neurons respond to stress by interacting with glucocorticoid receptors (GRs). GRs act in combination with NMDA receptors, a glutamate receptor essential for learning and memory. Together, they trigger a signaling pathway involving ERK-MAPK, a series of proteins that relay stress signals into the nucleus of neurons.
Inside the nucleus, this stress-induced signaling leads to epigenetic modifications: chemical tags on DNA and its associated histones that regulate gene expression without altering the genetic code itself. Specifically, stress causes two key changes on histone H3, and is demonstrated in the psychological stress pathway in Figure 1 below:
- Phosphorylation at serine 10 (S10)
- Acetylation at lysine 14 (K14)
This dual mark, H3S10p-K14ac, acts like a molecular switch, opening up tightly wound DNA so that genes can be transcribed. Which genes get turned on? Immediate-early genes (IEGs), like c-Fos and Egr-1, are crucial for synaptic plasticity and memory consolidation[1].
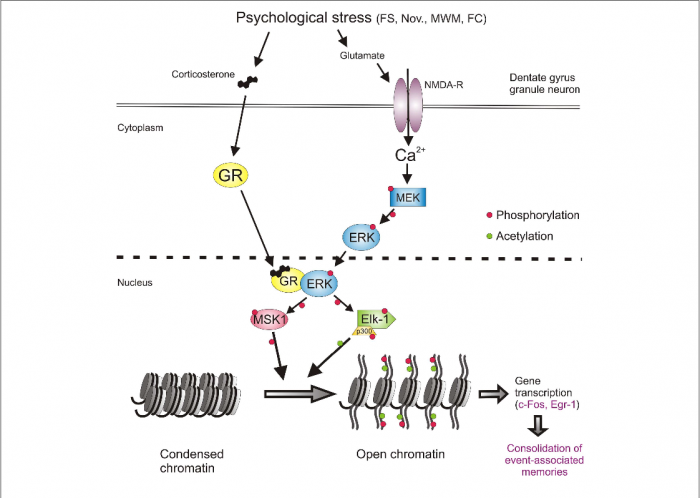
However, this epigenetic mechanism isn’t universal across the brain. While many regions activate c-Fos and Egr-1 during stress, only in the dentate gyrus (a subregion of the hippocampus) does their expression require H3S10p-K14ac[1]. This suggests that dentate gyrus neurons keep these genes inactivated under normal conditions, only releasing them when stress demands it.
What’s novel about this research is that it illustrates how glucocorticoids and glutamate signaling collaborate. Traditionally, steroid hormones like glucocorticoids were thought to act slowly, binding to receptors in the nucleus and modulating gene expression over hours. However, this research suggests that GRs physically interact with phosphorylated ERK to increase the activity of downstream kinases like MSK1 and Elk-1[1].
This interaction happens within minutes of stress, allowing glucocorticoids to amplify glutamate-driven signals and accelerate epigenetic modifications. Thus, GRs serve to amplify the ERK-MAPK pathway, ensuring the stressful memory is highly encoded.
Thankfully, the brain doesn’t let stress run unchecked. GABA, the brain’s primary inhibitory neurotransmitter, modulates the dentate gyrus’s response to stress through a baseline level of inhibition. This finding is supported by studies of drug interactions:
- Anxiolytics (like lorazepam, a benzodiazepine) suppress stress-induced H3S10p-K14ac and c-Fos activation.
- Axiogenics (like FG7142, a GABA-A receptor antagonist) do the opposite, increasing epigenetic and gene responses.
Even exercise plays a role. Long-term voluntary running in rats reduces anxiety-like behavior and dampens stress-induced ERK-MAPK signaling, likely by enhancing GABA synthesis[1]. This fits with real-world observations that exercise can help mitigate stress disorders[2].
Understanding these mechanisms has real implications for stress-related disorders like PTSD and anxiety. In vulnerable individuals, the balance between stress-induced memory formation and adaptive coping may go awry.
- Hyperactive GR/ERK-MAPK signaling: This might lead to the over-consolidation of traumatic memories (e.g. PTSD flashbacks).
- Dysregulated GABA control: Could result in excessive fear responses or failure to dampen stress reactions[1].
This also opens doors for potential therapies. If researchers can pinpoint drugs that target these epigenetic or signaling pathways (like MSK1 or Elk-1) there’s potential to disrupt maladaptive stress memories without affecting useful ones.
This research paints a vivid picture of how stress etches itself into our brains. It’s not just about neurons firing, it’s a combination of hormones, neurotransmitters, and epigenetic marks. While evolution likely designed this system to help us remember threats, sometimes it overshoots, leaving us stuck in loops of anxiety or trauma. The good news? The brain is extremely plastic. Exercise, therapy, and even pharmacological interventions can adjust these pathways, offering hope for better stress resilience.
Stressful events trigger glucocorticoids and glutamate signaling in the hippocampus, but the formation of long-term memories requires a precise epigenetic switch (H3S10p-K14ac) that activates memory-related genes. Therefore, understanding this mechanism reveals potential therapeutic targets for stress-related disorders like PTSD and anxiety.
References
[1] J. M. H. M. Reul, “Making Memories of Stressful Events: A Journey Along Epigenetic, Gene Transcription, and Signaling Pathways,” Front. Psychiatry, vol. 5, p. 5, Jan. 2014, doi: 10.3389/fpsyt.2014.00005.
[2] H. Alizadeh Pahlavani, “Possible role of exercise therapy on depression: Effector neurotransmitters as key players,” Behav. Brain Res., vol. 459, p. 114791, Feb. 2024, doi: 10.1016/j.bbr.2023.114791.