
Learning Beyond Memorization
Coming into this semester, I thought I had a solid understanding of what it meant to “learn” science. As a neuroscience major on a pre-med track, I’ve spent years studying pathways, mechanisms, and disorders. But this class challenged me to move beyond memorization and into something deeper—engagement.
One of the biggest shifts for me came from reading scientific papers and actually interacting with the concepts instead of just passively trying to understand them. At first, primary literature felt dense and overwhelming. But over time, I started to see patterns—how researchers ask questions, how they design experiments, and how they interpret results. More importantly, I began to connect those findings back to what I was learning in my other classes, especially biochemistry and neurochemistry.
That shift—from just learning content to actively engaging with it—was one of the most meaningful parts of this semester.
The Reality of Balancing It All
At the same time, this semester was not easy.
Balancing coursework, assignments, and everything else going on in my life was one of the biggest challenges I faced. There were moments where staying on top of deadlines felt overwhelming, and I had to be more intentional about managing my time and energy. This wasn’t just about academics—it was about learning how to show up consistently, even when I didn’t feel fully prepared.
In a way, this challenge was its own form of learning. It forced me to develop discipline, resilience, and a better understanding of how I work under pressure. These are skills that go beyond the classroom and will definitely carry into my future career.
Finding My Voice in Science
If there’s one area where I saw clear growth, it was in how I communicate science.
Through blog posts and artstracts, I learned how to take complex neurochemical and biochemical concepts and present them in a way that is accessible and engaging. Writing for a general audience pushed me to simplify without losing meaning, which is much harder than it sounds. It made me think carefully about what really matters in a concept and how to explain it clearly.
I also saw this growth during oral exams and class discussions. Being able to explain neurochemical concepts in conversation—with my professor and peers—was something I became more confident in over time. There’s something different about speaking your understanding out loud. It reveals what you truly know and where your gaps are.
Looking back, I’m proud of how much more comfortable I’ve become in these moments. Instead of feeling intimidated, I now see them as opportunities to think through ideas in real time.
Connecting Knowledge to My Future
As someone interested in going into healthcare, particularly in a patient-centered role, the skills I developed this semester feel very relevant.
Healthcare is not just about knowing information—it’s about communicating it clearly and compassionately. Patients don’t think in terms of biochemical pathways or signaling cascades. They need explanations that make sense to them, especially when they are already dealing with stress or uncertainty.
This class helped me practice that skill in a meaningful way. Whether I was writing a blog post or explaining a concept during an oral exam, I was constantly thinking about clarity, structure, and understanding.
I also developed a deeper appreciation for how interconnected different areas of science are. Taking biochemistry and neurochemistry at the same time allowed me to see how molecular processes translate into brain function and behavior. That kind of integrative thinking is essential in healthcare, where problems are rarely one-dimensional.
What Liberal Learning Means to Me
Before this semester, I understood liberal arts education as being “well-rounded.” But now, I see it more as being connected.
At Concordia, I’ve had the opportunity to explore different disciplines, but more importantly, I’ve learned how to see the relationships between them. This semester really highlighted that for me. Concepts I learned in biochemistry directly supported my understanding of neurochemical pathways, and together, they gave me a more complete picture of how the body works.
Liberal learning, to me, means being able to move between perspectives—scientific, social, and personal—and understanding how they inform each other. It’s not just about depth in one area, but about the ability to integrate knowledge across fields.
This approach has also encouraged me to think more broadly about real-world issues, especially in health and disease.
A Skill Worth Highlighting
If I were to highlight one skill I improved this semester on my resume, it would be:
Science Communication and Concept Integration
I’ve developed the ability to explain complex scientific ideas in a clear and accessible way, while also connecting concepts across disciplines. This includes writing for a general audience, engaging in discussions, and thinking critically about how different areas of knowledge overlap.
This is a skill that will be valuable not only in healthcare, but in any setting where clear communication and interdisciplinary thinking are important.
Understanding Metabolic Syndrome Through Multiple Lenses
One example that stands out to me in terms of interdisciplinary problem-solving is metabolic syndrome.
At first, metabolic syndrome might seem like a purely biological issue—something explained by insulin resistance, lipid metabolism, and hormonal regulation. And while those factors are important, this class helped me see that the problem is much more complex.
From a biochemical perspective, we can analyze pathways involving glucose regulation, fatty acid metabolism, and inflammation. From a neuroscience perspective, we can consider how the brain regulates appetite, reward, and stress responses. These systems influence behaviors like eating patterns and physical activity.
But we also have to think about psychological and social factors. Stress, mental health, access to healthy food, cultural dietary habits, and socioeconomic status all play a role in the development and progression of metabolic syndrome.
By integrating these perspectives, the problem becomes more complete. It also changes how we think about solutions. Instead of focusing only on medication or biological interventions, we can consider lifestyle changes, education, and community-level support.
This kind of interdisciplinary thinking reflects the goals of liberal learning – it allows us to approach complex issues in a way that is both informed and meaningful.
Growth That Extends Beyond the Classroom
Looking back, this semester was not just about what I learned, but how I learned.
I became more comfortable engaging with difficult material, more confident in expressing my understanding, and more aware of the connections between different areas of knowledge. I also learned how to navigate challenges and keep moving forward, even when things felt overwhelming.
These are lessons that go beyond any single class.
Moving Forward
As I reflect on this experience, I realize that learning at a liberal arts institution is not just about preparing for a career—it’s about developing the ability to think, connect, and engage with the world in a meaningful way.
This class pushed me to grow in those areas. It challenged me, but it also helped me see my own progress.
And while this may be the final blog post of the semester, the skills and perspectives I’ve gained will continue to shape how I approach learning in the future.
In that sense, this is not really an ending – it’s a continuation of the kind of learner I am becoming.
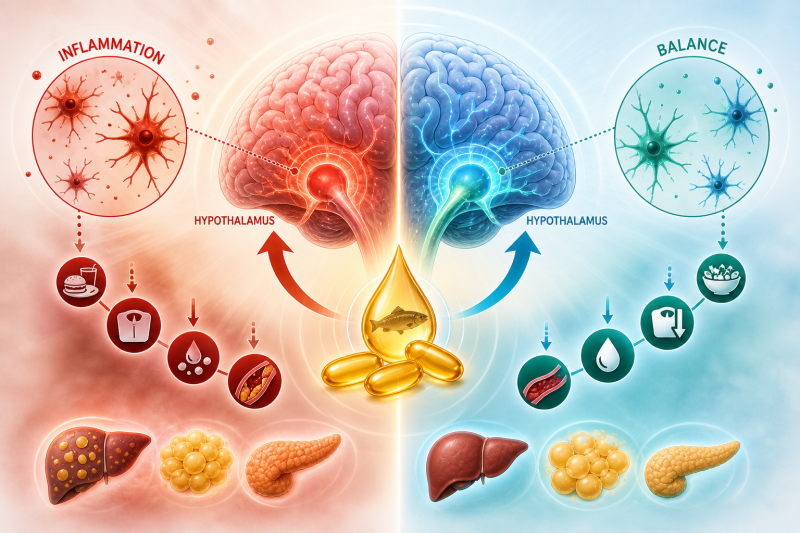
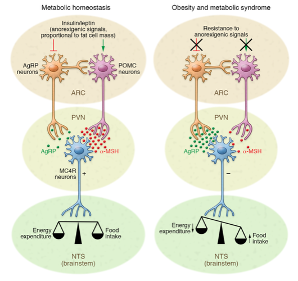 Figure 1: In metabolic syndrome, inflammation disrupts communication between hunger(AgRP) and satiety(MSH) pathways.
Figure 1: In metabolic syndrome, inflammation disrupts communication between hunger(AgRP) and satiety(MSH) pathways.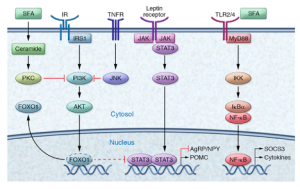 Figure 2(right): Omega-3s counteract inflammatory pathways that drive insulin resistance.
Figure 2(right): Omega-3s counteract inflammatory pathways that drive insulin resistance.
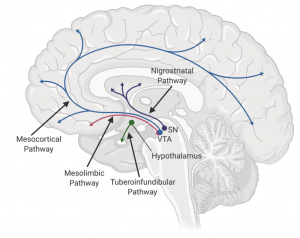 Figure 1(left): Dopaminergic pathways connecting key brain regions involved in reward and motivation. [1]
Figure 1(left): Dopaminergic pathways connecting key brain regions involved in reward and motivation. [1] Figure 2: Differences in dopamine signaling may shape how social interactions and repetitive behaviors are experienced in autism.[2]
Figure 2: Differences in dopamine signaling may shape how social interactions and repetitive behaviors are experienced in autism.[2]
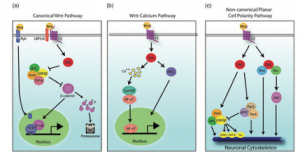
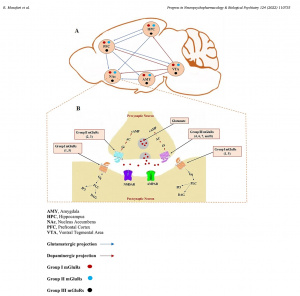
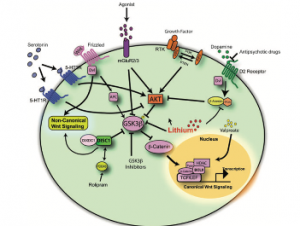 Figure 2 (left): Integration of dopamine, Akt/GSK3β, and Wnt signaling pathways converging on β-catenin-mediated transcription.[1]
Figure 2 (left): Integration of dopamine, Akt/GSK3β, and Wnt signaling pathways converging on β-catenin-mediated transcription.[1]






 A
A 

 Image sourced from
Image sourced from  image sourced from
image sourced from