Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder that impairs cognitive functions, specifically memory, making it the leading cause of dementia.[4] Despite not knowing the specific etiology and pathogenesis of AD, the hallmarks are amyloid-beta (Aβ) plaques and neurofibrillary tangles (NFTs).[1] Recent research has suggested that dysfunctional insulin signaling as well as neuroinflammation accelerate the progression of AD and its associated cognitive decline. Therefore, there is potential in targeting molecules involved in insulin’s signaling pathway including IDE, IRS, PI3K, mTOR, and GSK-3b for future treatment or prevention of AD.
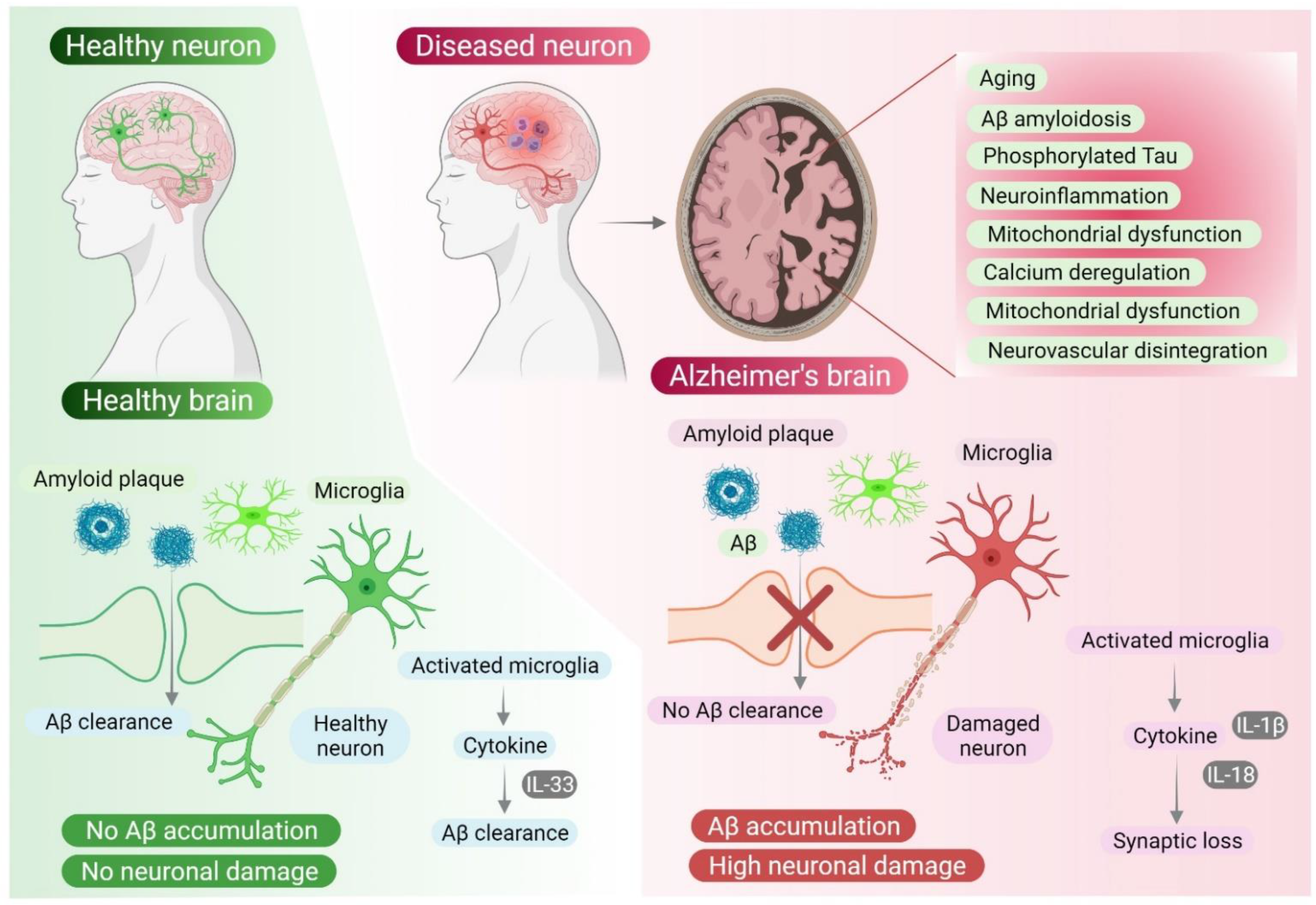
Figure 1. Two major hallmarks of Alzheimer’s disease: Aβ plaques and NFTs. [7]
Insulin Signaling – Why is it Important to AD?
Insulin, typically associated with glucose metabolism, has a regulatory role in the brain where it helps promote synaptic plasticity, neurotransmission, and neurogenesis. This is why insulin signaling is important and could be implicated in the progression of AD because when it is functioning properly improves cognitive function.
Insulin Resistance – A Possible Cause of AD?
Insulin resistance happens when the body becomes less responsive to the effects of insulin hormones. This can occur when there are lower levels of insulin binding to its receptors because insulin’s receptors can become desensitized. This can mean that even with insulin hormones being present, they might not bind to their receptors and activate their downstream pathways. Insulin resistance boosts oxidative stress, cytokines production (leading to neuroinflammation), and apoptosis. Insulin resistance leads to Alzheimer’s because it triggers a cascade of disrupted molecules in insulin’s signaling pathways, such as IDE, IRS, PI3K, mTOR, and caspases (Nrf2 and NF-kB).

Figure 2. Role of the insulin signaling pathway (insulin and its various molecules) in neurodegeneration and Alzheimer’s disease. [5]
Amyloid-Beta to Aβ Plaques
Amyloid-beta peptides are cleaved from the glycoprotein amyloid precursor protein (APP) that plays a major role in neuronal development and signaling. Amyloid-β plaques occur when amyloid-beta proteins clump together outside of the cells. When plaques build up they may block cell-to-cell signaling at synapses or activate an immune response that causes excess cytokines to be released and may be a leading contributor to neuroinflammation in AD cases.
Figure 3. Formation of Amyloid-β plaques from the precursor APP protein. [3]
Tau Proteins to Neurofibrillary Tangles
In healthy neurons, tau proteins help to stabilize and coordinate microtubules, which help give neurons their structure. When insulin signaling is disrupted and PI3K pathways are not being activated properly, Akt does not phosphorylate GSK and inactivate it. When GSK is active it phosphorylates the tau proteins, causing them to break away from the microtubules and clump together into NFTs. Neurofibrillary tangles accumulate inside the cell and block neuron’s normal functioning, including the synaptic communication between neurons. Tau proteins leaving microtubules could also play a role in neurodegeneration because microtubules would not stabilize the cell structure and neuron extensions.
Figure 4. Formation of NFTs from tau hyperphosphorylation. [2]
What Goes Wrong in Insulin Signaling Pathways?
IRS
The insulin receptor substrate relays the signal from the insulin receptor to activate the downstream responses. However, in Alzheimer’s IRS-1 is downregulated, which negatively affects the PI3K pathway. Reduced activation of PI3K/Akt enhances the activation of GSK-3b which increases tau phosphorylation and causes the formation of NFTs.

Figure 5. Insulin signaling pathway shown simplified to highlight IRS activation of PI3K pathway. [9]
PI3K and mTOR
Insulin resistance disrupts the PI3K signaling pathway, impacting the activation of mTOR. Since the PI3K pathway activates the mTOR gene to produce nutrients, proteins, and lipids that help other cells grow and create a healthy environment. In AD, with the PI3K pathway not activating mTOR, mTOR can’t produce nutrients to help insulin grow and produce more cells, which creates a bad cycle.
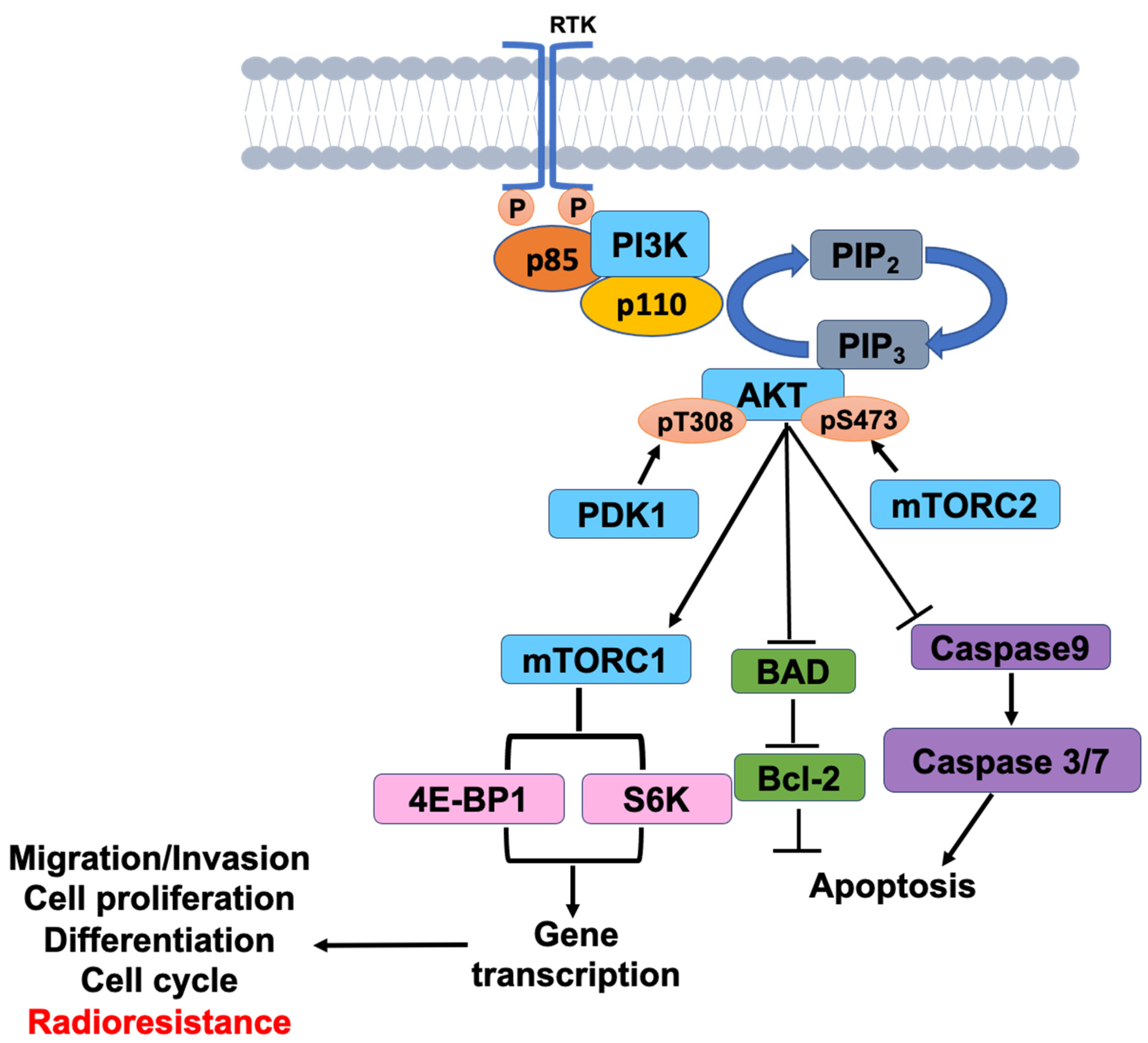
Figure 6. PI3K signaling pathway activation of the mTOR pathway involved in cell growth. [8]
IDE
IDE is an enzyme that breaks down insulin and the amyloid-beta protein. In AD, hyperinsulinemia (too much insulin) and insulin resistance affect the activity of IDE, causing insulin to compete with amyloid-beta to bind to the enzyme IDE. When insulin resistance occurs, lower levels of IDE happen because there is a downregulation of the PI3K signaling. Both reduce the clearance and degradation of amyloid-β in the brain, leading to the accumulation of Aβ plaques.

Figure 7. Insulin decreases the IDE-mediated beta-amyloid (Aβ) degradation through competitive inhibition. Insulin resistance also plays a role as it lowers the IDE expression and decreases IDE-mediated Aβ degradation. [6]
Caspases (NF-kB and Nrf2)
Disrupted insulin signaling (or insulin resistance) activates NF-kB, a proinflammatory transcription factor that releases cytokines and contributes to neuroinflammation, further impacting the progression of AD.
Nrf2 is a regulatory transcription factor that is activated in response to oxidative stress, promoting antioxidation. However, in Alzheimer’s Nrf2 levels are decreased, leading to an increase in insulin resistance and oxidative stress.