Neurochemistry has allowed me to integrate and apply the skills and competencies I have gained throughout my time at Concordia in the following ways. It caused me to test my knowledge thus far in Neurochemistry and beyond, and contribute, communicate, and deliver it effectively to my peers. While scientists are often able to understand and conceptualize material for themselves, they often lack the ability to communicate it effectively and cohesively to others. This course challenged and tested me in this area, as we were tested on our ability to not only know the science, but also effectively communicate it as well.
Neurochemistry has allowed me to take my love for learning, especially learning neuroscience, and excel in it. The course dove deep in the mechanisms and signaling in many different disorders, and I was able to practice and deepen my love for learning by examining and broadening my scope of understanding around these disorders, which helped me to become even more passionate about the ability to heal or change the signaling mechanisms.
Throughout all of my educational career, I have taken courses all over the “science spectrum.” Courses that include biology, chemistry, neuroscience, morality, ethics, math, etc. All of these courses feel combined and put to the test in Neurochemistry. All of these different types of courses matter when it comes to neurochemistry and background in all of them was very important. This knowledge allowed me to have a firm foundation in my education as a whole, and transfer this knowledge into the Neurochemistry course.
The course itself integrates all areas of learning. Educationally, personally, and professionally, by testing your knowledge and ability to apply that to a classroom setting, and in the community as well. This integration is important for “BREWing” as it allows a student to take the skills and knowledge they have learned and apply it in the real world. I think the course was a set and up and reflective of this very idea, as it allows the students to engage with others and share their insight and knowledge, as well as personal experiences with the class. This was great to see as we all have different backgrounds and insights to offer, and we can all learn something from one another through sharing and collaborating together. Since the course was based on discussion and communicating the science, this is a perfect example of the skills that can be applied to real world situations when we as students BREW. We will need to effectively communicate and collaborate with others about the science, about our own experiences and perspectives as well as justify and reason, which the course design allowed for. Overall, the Neurochemistry course allowed me to grow in my knowledge of science, and ability to effectively communicate this knowledge with others.
To me, learning at a liberal arts college is meant to produce well rounded individuals that are as prepared as possible to take on the problems of the world as possible. Through this, a variety of classes and disciplines are required to be taken by students to ensure they are exposed to different studies, ideas, and careers. But why should a chemist have to take religion classes? Or a theatre major having to a lab science class? Despite the two examples mentioned above seeming very opposite at face value, they are more interconnected than many think. This is shown through a quote from Thucydides, who was perhaps the most important historian of ancient times as he accounted for the nearly three-decades long conflict between ancient Sparta and Athens as well as the war between ancient Greece and Persia:
“The society that separates its scholars from its warriors will have its thinking done by cowards and its fighting by fools.”
This quote shows in dramatic fashion the importance of interdisciplinary study. But take a prelaw major for example. How could we expect a future environmental lawyer to be effective if they’ve never been in a biology class? So, while liberal learning might not always be the most clear to students being required to take the classes, there is a deeper connection between different disciplines.
Figure 2: A man clearly confused by the problem ahead of him.
How my resume improved
College is all about building the skills necessary to be successful in the future, whether that be in life or through a career. This semester, perhaps more than any other, had me looking like the poor guy in Figure 2. Between this Neurochemistry class and Physical Chemistry there were countless times where I simply had no idea how to answer the question at hand, whether it be derive the Maxwell relations or determine what is causing an increase in prevalence of PTSD and other stress related disorders in women compared to men. But, it was through questions like these that I really developed my ability to problem solve. Of course everyone is going to put that they work well independently and are a good problem solver on their resume, but what if your problem has infinitely many wrong answers, but also an answer that isn’t necessarily right or solvable?
I think that is the kind of problem solving I found myself really developing this semester, especially with the way the exams were designed. By not having the paper and only some of the data coming from it, we as students had to use our prior knowledge of signaling pathways to derive a viable cause for the data we were given. During both of these exams, I found myself reading through all the given information and then just thinking ‘I have no idea what’s going on.’ But I just kept piecing together the things I did know with what was given and soon enough I had a pretty decent guess at what was happening in the article.
What the future holds
The skills learned in this class as well as the ones I’ve developed during my Concordia career, I believed have given me a solid foundation to work off in the future. As I mentioned earlier, this class really helped me problem solve through situations that don’t have a clear answer. I think this will be very helpful as I am planning to attend graduate school next fall. In that environment, I will be working through problems that don’t have an answer yet. With that, I will need to be able to effectively communicate my ideas and findings to other people. While blogging is a fairly casual way of doing that, the skill still helps me be able to communicate with other effectively. And because of these reasons I believe that this class as well as Concordia College’s liberal learning program has helped to prepare me to be successful in the future.
I had expectations of this course before taking it but was surprised in the end. My expectations of this course included having lectures, exams, and quizzes similar to the other courses I have taken. The format of this class was a new experience for me and I was hesitant at the start of the semester about how it would go. I was pleasantly surprised about how much I enjoyed and learned throughout this class. The process of learning was very independent during neurochemistry. I learned more skills related to finding information on my own and communicating it to classmates. The topics we discussed required researching them independently and then teaching them to other students. I acquired new skills about how to communicate information efficiently and effectively.
The skills I learned in this class will greatly benefit me in my future career. I plan to work for a few years and then apply to medical school. Learning how to conduct independent research, properly read research articles, and successfully communicate knowledge to others are all skills I expanded on throughout neurochemistry. These are all necessary skills to be successful in any career, especially in the field of medicine. I believe my 2.5 years at a liberal arts college have helped me realize the capabilities I have. There have been many opportunities for me to learn about the topics I want to and teach others about them. Attending a liberal arts college, to me, means providing students with a well-rounded education and preparing them for life after college.
If I were to highlight my resume with skills that I improved upon this semester, I would choose to discuss how I have gotten better at reading and understanding research articles. There are an endless number of topics to read articles about, but how you read them is important. It is difficult to understand all topics within a paper if you do not have any previous knowledge of the research being discussed. Neurochemistry has helped me to start reading every paper with an open mind and knowledge that I will not understand everything after reading it one time. It is important to note questions and the need for clarification when reading an article. I enjoyed this class because I was able to openly ask questions and talk about things I did not understand. This course helped me to effectively read research articles and build communication skills when discussing the article.
I was able to integrate a lot of information from previous courses into this class. I have taken a variety of biology, chemistry, neuroscience, and psychology courses that provided relevant information for neurochemistry. Previous courses have focused on different skills such as exam taking, dissections, and working in a laboratory. All things I learned in the past were applicable to different weeks of content during neurochemistry. I felt I had a greater understanding of topics such as mental illness, anxiety, and sleep because of my previous courses in psychology and neuroscience. It was interesting to learn more about topics I did not know much about such as autism, obesity, and endocannabinoids. Overall, I enjoyed this course and how I was able to integrate old skills into expanding upon new ones.
As a student who is a psychology major and does not have a knack for chemistry, I was very intimidated going into this class. I was more focused on the idea that this would be the final core class I would have to take at Concordia, so I started off rough in the class. I was less focused on understanding and applying the material to other things I had learned and more focused on going through the motions to get through the class. After the first exam, I had a moment of realization; I knew that I was not doing enough in order to be successful, and I hated the idea of that. From that point on, I was determined to learn the material properly. I had continuously told myself that I just “wasn’t good at chemistry so I won’t do well.” That fixed mindset was what was weighing me down, and after this class, I now believe in myself when it comes to chemistry. My biggest learning achievement was learning how to properly read scientific articles. When I first started the class, I would read the articles but not get much information out of it. It was almost like I didn’t know what I was supposed to be getting out of the article. Once I changed the style in which I read the articles, it opened a whole new door of understanding for myself. I started to enjoy learning the material, I would go out of my way to do extra research for different areas of the article, I had re-instilled my love for learning. The biggest application this class had to offer for me was communication with other students. I tended to be quiet in class considering I didn’t know much about the material compared to other students. Over time though, I started to realize it was less about knowing the material and was more about being able to voice your thoughts with the other students. Communication was my most improved skill by far as a result of this class. I found myself participating more in not only discussions for this class, but also in my job and other classes as well. I learned to be more confident in what I say which made me more likely to speak up. Learning at a liberal arts institution is so much more than just learning class material. To me, it’s about applying what you learn to what you do, how you can make people’s lives better around you with the information provided. It’s about the lessons you’ll carry into your next chapter and learning there’s no limit on what you can accomplish. The problem that required the most disciplinary perspective was the PEAK project. This is because there are students from two entirely different disciplinaries trying to work together as one instead of two. Groups can be so focused on the fact that their disciplinaries are different, like working from a social work perspective compared to a chemistry perspective, but what’s great about having two different groups is that you can use multiple disciplinaries to your advantage. The social work students were able to use the skills they’ve learned from classes to properly engage and spread the word of the project while the chemistry students were able to offer the proper material for putting together an effectively worded project. When two groups can come together, the work can be inspirational; when two groups can’t come together, there is not hope for a successful project. It’s easy to think that because other people are different from you, that you won’t be able to work together. However, I think that was the lesson Concordia instilled on us best, that everyone is different and no matter how different you are from someone, the only thing that is stopping you from working well together is the thoughts in your own mind.
You feel grass tickling your neck and arms. You open your eyes, but the people standing around you all of sudden seem about 10 feet tall as they lean over you. Something is not right, and you realize you are laying on the ground on your back. You have been hit.
Thinking back to the progression of your day, you got up and went to school in the morning, then got to PE class, where you all played soccer. Your head met your friend’s knee with an excessive force that sent you straight to the ground. Now you are being told the words “you got a concussion”… but what now?
A recent Gallop poll found 49% (up from 45% in 2017) of Americans have tried marijuana in some form at some point (Hubbard, 2021). Some are doing it for health benefits, others for recreation. Whatever reason people are using marijuana, it is clearly an important topic to discuss based on the sheer amount of people using it in the USA alone. To learn more about the stats and history of marijuana in the US, check out this link! There are some interesting health benefits to some of the compounds in marijuana called cannabinoids. The two most well known and studied are THC and CBD. At this point in time, these chemical abbreviations are nearly household names! In this post, we will explore some of the health benefits of CBD and THC are, as well as explaining the science behind why these chemicals can be so effective.
Is Cannabis Really “Naturally” Found in the Brain?
Yes. No. Well, sort of! The brain produces a class of chemicals called endocannabinoids. these have names like anandamide, arachidonylethanolamine, and 2-archidonoyl glyerol. These are endogenous cannabinoids, similar to form and function to THC and CBD. Fun fact, anandamide is derived from the Sanskrit word “ananda” which means internal bliss (Scherma, 2019). The Sanskrit meaning hints at some of its functions in modulating brain chemistry.
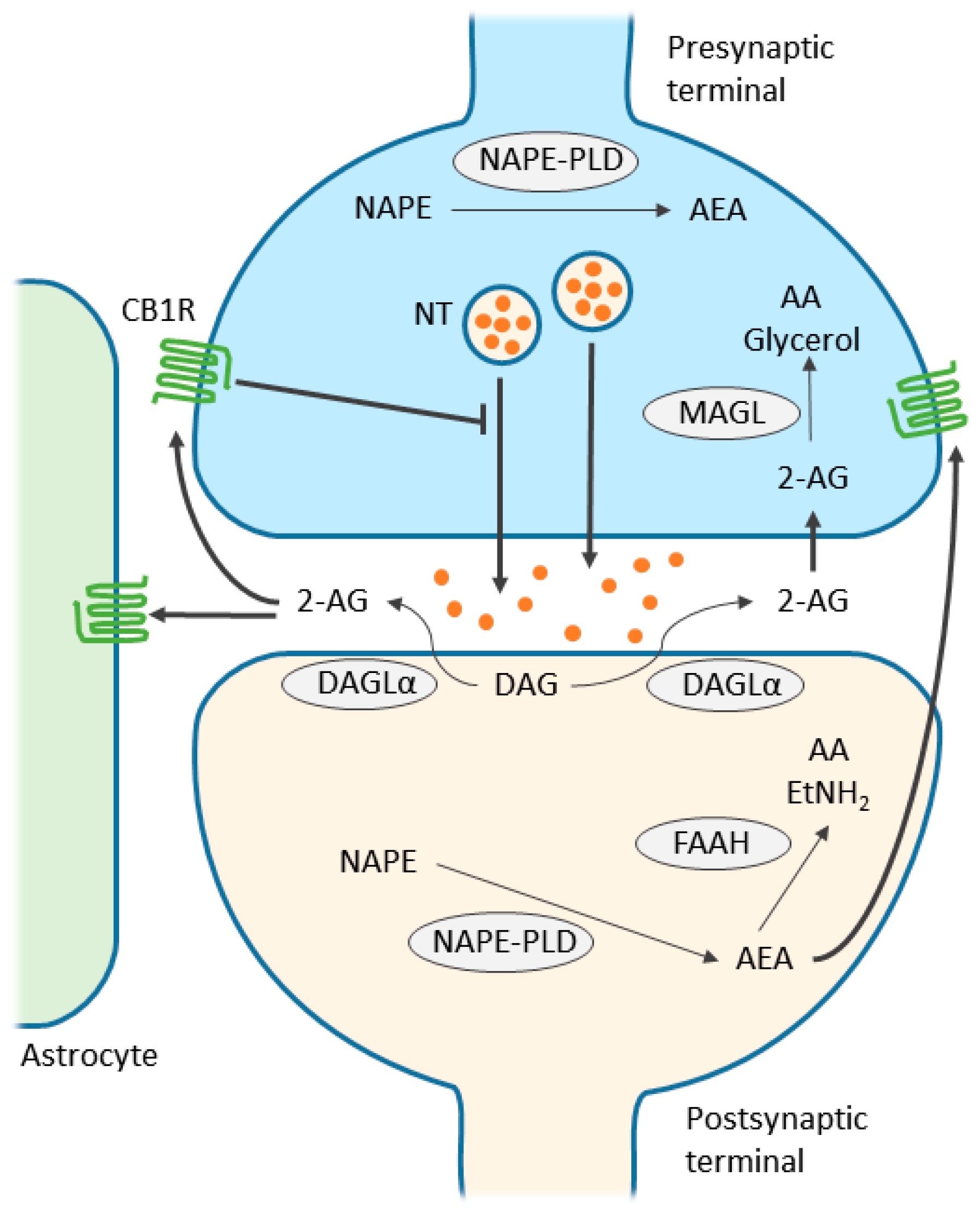
Interesting! But where do these endocannabinoids come from? Interestingly, they are modified from fat in your cell membranes. Neuronally, this occurs in the post-synaptic neuron, AKA the neuron being being stimulated. See figure 1 below for a nice visual.
Figure 1. Reproduced from Zou et al. This figure shows endocannabinoid signaling between the pre-synaptic neuron (blue) and the post-synaptic neuron (green).
So great. We know the names of several endocannabinoids, but they have to bind somewhere to exert any of their effects. Endocannabinoids bind primarily to two receptors: CB1 and CB2 (Kendall, 2017). CB2 receptors are mostly found in the immune system, and when activated by endocannabinoids, inflammation levels increase and drug self-administration frequency decreases (Kendall, 2017). On the other hand, CB1 receptors are expressed throughout the body (Kendall, 2017) and are responsible for many of the positive benefits and side effects of THC and CBD.
Health Benefits of THC and CBD
The claims made online about what THC and CBD can do to a person range anywhere from giving one brain damage, to curing any ailment. Unfortunately, because because marijuana is a schedule I drug, conducting research on it is very difficult and makes a lot of these claims unsubstantiated. Because of this, I will focus on only a few of the positives of cannabinoids that I feel are substantiated with at least some rigorous, scientific evidence.
Should Grandpa Be Toking Up?
An interesting study conducted in mice found that older mice given THC had improved memory (Sarne, 2018). Not only did these old THC mice have the memories that matched young mice, but so did the expression levels of genes involved with memory (Sarne, 2018). This suggests that THC administration could be a viable way to slow or even reverse aspects of aging in the brain (Sarne, 2018).
What About CBD?
When one reviews the literature on CBD, it becomes apparent that it’s anti-inflammatory properties are one of it’s greatest assets in treating disease. A randomized clinical trial (RCT) found CBD is effective at significantly reducing the pain caused by arthritis (Verrico, 2020). Another study found that CBD can help treat Chron’s disease, and even wean some people off of steroid therapy (Naftali, 2013). Yet another RCT found CBD to be effective in preventing inflammation driven hyperpermeability of the human gut (Couch, 2019). The connection between all these studies is the anti-inflammatory effect of CBD. Too much inflammation can lead to many different diseases, and CBD seems like an effective tool to combat excess inflammation.
Conclusion
The amount of Americans using marijuana has been increasing ever since the records began. Because it is a schedule 1 drug, research using marijuana is very difficult and there are many questions science can answer, but isn’t being allowed to do so. Nonetheless, some promising studies have been conducted and have found health benefits for both THC and CBD. From the memory enhancing and anti-aging effects of THC, to the anti-inflammatory benefits of CBD, cannabinoids offer hope for treating a variety of ailments.
Sources
Couch, D. G., Cook, H., Ortori, C., Barrett, D., Lund, J. N., & O’Sullivan, S. E. (2019). Palmitoylethanolamide and Cannabidiol Prevent Inflammation-induced Hyperpermeability of the Human Gut In Vitro and In Vivo-A Randomized, Placebo-controlled, Double-blind Controlled Trial. Inflammatory bowel diseases, 25(6), 1006–1018. https://doi.org/10.1093/ibd/izz017
Hubbard, K. (2021). Record high: More Americans are trying marijuana, Gallup … Record High: More Americans Are Trying Marijuana, Gallup Poll Finds. Retrieved December 9, 2021, from https://www.usnews.com/news/national-news/articles/2021-08-17/record-high-more-americans-are-trying-marijuana-gallup-poll-finds.
Kendall, D. A., & Yudowski, G. A. (2017). Cannabinoid receptors in the central nervous system: Their signaling and roles in disease. Frontiers in Cellular Neuroscience, 10, 294. https://doi.org/10.3389/FNCEL.2016.00294/BIBTEX
Sarne, Y., Toledano, R., Rachmany, L., Sasson, E., & Doron, R. (2018). Reversal of age-related cognitive impairments in mice by an extremely low dose of tetrahydrocannabinol. Neurobiology of aging, 61, 177–186. https://doi.org/10.1016/j.neurobiolaging.2017.09.025
Scherma, M., Masia, P., Satta, V., Fratta, W., Fadda, P., & Tanda, G. (2019). Brain activity of anandamide: a rewarding bliss?. Acta pharmacologica Sinica, 40(3), 309–323. https://doi.org/10.1038/s41401-018-0075-x
Naftali, T., Bar-Lev Schleider, L., Dotan, I., Lansky, E. P., Sklerovsky Benjaminov, F., & Konikoff, F. M. (2013). Cannabis induces a clinical response in patients with Crohn’s disease: a prospective placebo-controlled study. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association, 11(10), 1276–1280.e1. https://doi.org/10.1016/j.cgh.2013.04.034
Verrico, C. D., Wesson, S., Konduri, V., Hofferek, C. J., Vazquez-Perez, J., Blair, E., Dunner, K., Jr, Salimpour, P., Decker, W. K., & Halpert, M. M. (2020). A randomized, double-blind, placebo-controlled study of daily cannabidiol for the treatment of canine osteoarthritis pain. Pain, 161(9), 2191–2202. https://doi.org/10.1097/j.pain.0000000000001896
When I first signed up for neurochemistry, I was worried about my lack of knowledge in regard to chemistry, but I knew I had a love for neuroscience that I wanted to learn more about. Concordia has given me the opportunity to take what I love and learn about it in more ways than one. While taking other neuroscience classes here I have learned the basics and lightly touched on different pathways that are involved. However, it was never to the extent of what neurochemistry had to offer, which is understandable to have a class fully designated to looking at all different signaling pathways and their correlations to many different areas in the brain. I knew I would struggle with my lack of chemistry knowledge but with the help of many classmates I became more familiar with the information that was given during class. Having a good background in chemistry is important for this class but do not think that your lack of information will make your opinion less than that. Everyone comes into the class with knowledge about different topics and experiences that aid in conversations regarding every topic. When thinking about this struggle, Concordia’s motto of BREW was engraved into my mind. We as Concordia students base our four-year educational experience from our rooted value being BREW. Becoming responsibly engaged in the world is a way of life. It’s looking at the world and understanding the problems that each person may face, helping always, and stepping forward when everyone else might be afraid. The experiences and lessons I learned in this class taught me how to engage in my world, while also showing me how I could help others from the lessons I learned. This class has also deeply enforced my love for learning, especially when it comes to something that I may not feel the most confident in. It has made me reach out of my comfort zone when asking other students for clarification and well as showing me different study methods that are more beneficial than what I had been using previously. For example, previously I would look over the topic at hand and try and memorize what was being discussed. However, this was not the most efficient method for this class, instead, I found it better to draw out the different signaling pathways in order to see what exactly they followed and how different proteins interacted within that pathway. The types of learning that were involved in neurochemistry highly centered around group work/discussion. For this type of class I think this is the most beneficial way in which the subject can be taught. If we were to sit down and have the class purely lecture based, I feel as though it would not be as enjoyable. Though it is important to understand the baseline of what the differentarticles are talking about we would miss all the questions that we as individuals have on varying topics. This also teaches us how to question what we are reading and find ways to answer the questions we have through the use of published research. If I were to highlight a skill this class has provided me with would be critical thinking, how to ask questions and find the answers on your own.This way of learning is just another way in which liberal art institutions like Concordia College seek to benefit their student more than other institutions. We are able to ask questions and seek out different ways to solve them. Even more so, students that took part in this semester peak project were able to conduct research on a given topic and see how our local area is affected by this. Many of us were able to give back to the community in so many ways and help those that are in need. That is also something I love about Concordia College. We aren’t just a school focused on good grades and going through the motion of life, we are an institution focused on seeing problems within our community and reaching out a helping hand. This way of problem solving/helping will be instilled in all of us for years to come.
Childhood-onset schizophrenia (COS) is very rare. It is only considered to be COS when diagnosed in children under 13, but full onset of the disease often does not occur until late adolescence or early adulthood. Although, if a child is experiencing hallucinations, delusions, or psychosis (more of which we will get into later) this can definitely be a cause for concern. It is important to understand that many children display unusual behavior; their brains are not even near to being fully developed! Also, before jumping the gun on schizophrenia, these symptoms can point to other psychiatric disorders such as depression, anxiety, and autism. A common worry that I have seen for parents is a child’s development of an imaginary friend. Is it an hallucination, a delusion, or simply a child with a healthy imagination?
What are Imaginary Friends?
Imaginary friends, also known as imaginary companions (ICs), have been seen throughout adolescent’s lives for years. It is by far not an uncommon phenomenon. Research even goes so far as to show that 65% of children up to age 7 experience an IC (Taylor et al., 2004). There are multiple reasons why adolescents may develop an IC:
Each of these reasons may be considered beneficial to a child’s development, rather than detrimental (Figure 1). Research has shown that benefits may include advanced social cognition, healthy coping strategies, superior emotional intelligence, and especially heightened creativity (Majors & Baines, 2017)! Imaginary friends can take any form that may be important to the child, whether that be a person, animal, or stuffed animal. More likely than not, ICs are a healthy aspect of child development, and it is important to understand the difference between imaginary friends and schizophrenic hallucinations.
Hallucinations and Delusions in Schizophrenia
Often times, parents either disregard the presence of an imaginary friend, or discourage the act altogether, due to worries in their child’s perception of reality (Majors & Baines, 2017). Although, as described above, ICs differ greatly from symptoms seen in schizophrenia such as hallucinations and delusions. Hallucinations can come in many different forms, including visual (sight), auditory (hearing), olfactory (smell), and tactile (feelings). In the case of schizophrenia, the individual does not understand these hallucinations are fake, and they are not in control. In children with imaginary friends, they most often understand that their friends are not real, and the child is in control of them (Fritz, 2015). Delusions are beliefs that do not align with reality. These include beliefs that someone is planning to harm them, beliefs that one is ill, beliefs that someone is in love with them, or beliefs that one is superior to others. Again, this is far different from a child’s development of an imaginary friend, as they are not out of touch with reality.
When to Take Action
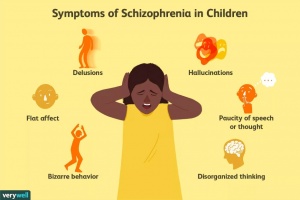
A parent should become concerned if their child beings displaying these types of behavior (Fig. 2).
Only speaks when spoken to; replying with short answers
Flat, monotonous facial expression
Beyond this, it is important to understand that schizophrenia in children is very rare. It is also a disorder of development, so major symptoms often do not show until late adolescence or teen years. Also, many of these symptoms correlate with other psychiatric illnesses that are not quite severe as schizophrenia, and often enough, they are just “symptoms” of being a developing kid. Although, if a parent is still concerned, it can never hurt to get a check-up!
Article Summary
Schizophrenia is a developmental disorder with a variety of positive and negative symptoms. The article focuses specifically on the role of the Wnt signaling, GSK3ß, and dopamine release. When there is an increase in dopamine, this inhibits Akt, leading to an increase in activation of GSK3ß. In regard to Wnt signaling, increases in dopamine also inhibit TCF/LEF-mediated transcription through the direct sequestering of ß-catenin. A primary treatment for schizophrenia, lithium, acts through the direct inhibition of GSK3ß, leading to an accumulation of ß-catenin and therefore, TCF/LEF-mediated transcription. There are also multiple genetic factors involved, primarily DISC1. DISC1 has been shown to inhibit GSK3ß, but it is dysfunctional in schizophrenia. Through this article, it appears as though the inhibition of GSK3ß, a decrease in dopamine release, and an upregulation of ß-catenin activation and TCF/LEF-mediated transcription are plausible targets for schizophrenia treatment (Singh, 2013).
References
Fritz, G. K. (2015). Imaginary friends . CABL, 31(5). https://doi.org/https://doi.org/10.1002/cbl.30041
Majors, K; Baines, E; (2017) Children’s play with their imaginary companions: Parent
experiences and perceptions of the characteristics of the imaginary companions and purposes served. Educational and Child Psychology , 34 (3).
Singh, K. K. (2013). An emerging role for Wnt and GSK3 signaling pathways in Schizophrenia.
Taylor, M., Carlson, S. M., Maring, B. L., Gerow, L., & Charley, C. M. (2004). The
Characteristics and Correlates of Fantasy in School-Age Children: Imaginary
Companions, Impersonation, and Social Understanding. Developmental Psychology, 40(6), 1173–1187. https://doi.org/10.1037/0012-1649.40.6.1173
Post-traumatic stress disorder (PTSD) is a mental health disorder that develops after witnessing or experiencing a traumatic event. It has been found that memories of these traumatic events are often much stronger than the formation of non-traumatic memories (Reul, 2014). During the experience of traumatic events, the body enters fight-or-flight mode in which many normal body functions are halted, including working (short-term) memory. Due to this, the body becomes extremely vigilant, absorbing as much as they can about their current surroundings. The brain attaches smells, feelings, visuals, sounds, etc. to this event, storing them in long-term memory. Since this event is unable to be fully processed, the brain may cause recurring bouts of anxiety, known as triggers. These triggers are due to a combination of three memory processes:
Strong perceptual priming (exposure to a specific stimulus evokes a strong response to another stimulus).
Strong associative learning (strong relationship between two stimuli).
Poor memory elaboration (decreased ability to enhance an existing memory with new information) (Ehlers, 2015).
All of these processes are manifestations of underlying genetic and molecular changes in response to stressful events. Some of these changes include activation of the MAPK signaling pathway in the brain and subsequent gene transcription (c-Fos and Egr-1) that enhance traumatic memory stability (Reul, 2014).
Types of PTSD Triggers
Triggers are reactions that make one act as if their body is in danger, even though they harmless themselves. These can be experienced in many different ways:
Panic attacks
Figure 1
Dreams or vivid memories
Violence or aggression
Increased startle response
Substance abuse
PTSD triggers may be either internal or external. Internal triggers are those that one experiences within their body such as anger, pain, increased heartrate, muscle tension, and loneliness. External triggers are people, places, or things that one may encounter throughout their day. These include an anniversary date, a certain place, a movie or television show, a specific smell, or a person that serves as a traumatic reminder. Essentially, anything that reminds an individual of their traumatic experience may serve as a trigger.
Recognizing PTSD Triggers
It is important for one with PTSD to identify what their triggers may be in order to seek proper treatment. Due to the various reactions one may have to their triggers, and the amount of possibly triggering experiences, identification may be very difficult. It becomes especially difficult with sensory triggers, such as taste, smell, or touch. In order to determine one’s triggers, it is important to ask questions such as, “Where and when was I when my symptoms flared up?” or, “What was my experience during this flareup?”. Although, the primary and most beneficial way to discover one’s triggers is discussion with mental health professional.
Interestingly enough, after identifying one’s triggers, repeated exposure to them is one of the most effective treatments, also known as prolonged exposure therapy. This allows one to remove that trigger from the traumatic experience, bringing it to the present where it no longer holds significance. Along with this, there are a variety of other coping mechanisms one may pursue:
It is also imperative to stray away from unhealthy coping mechanisms, such as alcohol and drug use. The experience of PTSD triggers is a difficult and challenging one to overcome, but understanding their development, recognizing one’s triggers, and seeking help are the most important steps one can take.
References
Ehlers, A. (2010). Understanding and treating unwanted trauma memories in posttraumatic stress disorder. Zeitschrift Für Psychologie / Journal of Psychology, 218(2), 141–145. https://doi.org/10.1027/0044-3409/a000021
Reul, J. M. (2014). Making memories of stressful events: A journey along epigenetic, gene transcription, and signaling pathways. Frontiers in Psychiatry, 5. https://doi.org/10.3389/fpsyt.2014.00005
The endocannabinoid system is a complex biological system that helps to regulate various processes in the central nervous system. The endocannabinoid system plays a role in pain, memory, neuroprotection, and modulation of synaptic plasticity. The endocannabinoids are endogenous ligands that bind to CB1 and CB2 receptors in the brain and are produced via enzymatic degradation of naturally-occurring molecules. In English, this basically means that the endocannabinoids are produced by molecular scissors that cut off certain pieces of molecules that are already existing in the body. These newly-cut molecules then bind to the same receptors that are activated when one ingests marijuana. The endocannabinoids are responsible for regulating many things throughout the body, so I think it is important to discuss how endocannabinoid signaling works.
Endocannabinoid Signaling:
Endocannabinoids operate as retrograde messengers, meaning they mediate function on the pre-synaptic cell, rather than the post-synaptic cell. Endocannabinoid signaling begins the same way all neuronal signaling does—an action potential is sent through the neuron and a neurotransmitter is released. The neurotransmitter then binds to the post-synaptic cell, which allows for the influx of calcium into the post-synaptic cell. This is where endocannabinoid signaling differs from most other types of signaling. When calcium enters the post synaptic cell, endocannabinoids are released into the back into the synapse and bind to the CA1 and CB2 receptors (higher affinity for CA1 receptor, though). Once the endocannabinoids bind to their receptors, they can induce one of two things: depolarization-induced suppression of inhibition (DSI) or depolarization-induced suppression of excitation (DSE). The endocannabinoids induce DSE and DSI the same way, but which is induced depends on the type of pre-synaptic cell.
Depolarization-Induced Suppression of Inhibition
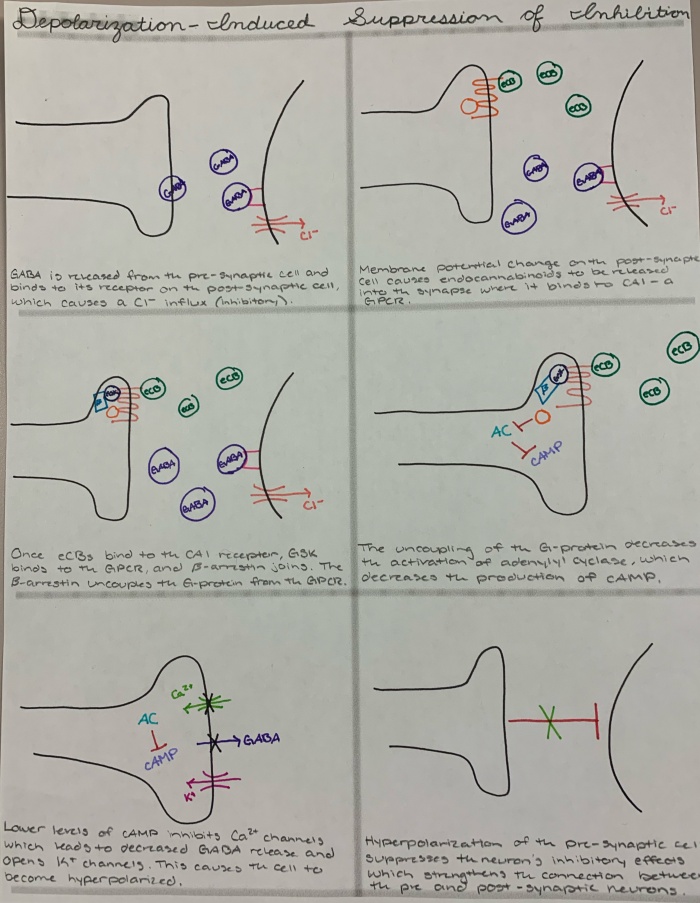
DSI is one result of endocannabinoid signaling. The CB1 and CB2 receptors are G-protein-coupled receptors (GCPR), which means they are associated with a G-protein. When the endocannabinoids are released from the post-synaptic cell and bind to the CB1 receptor, a complex signaling cascade occurs. The GPCR becomes a substrate for another enzyme called G-protein-coupled receptor kinases (GRK). GRKs are then targeted by beta-arrestin, which decouples the G-protein from the GPCR. Because the G-protein is uncoupled from the receptor, adenylyl cyclase is not activated, which then correlates with a decrease the levels of cyclic AMP (cAMP). The lowered levels of cAMP the inhibit calcium channels, thus decreasing the levels of neurotransmitters that are released, which opens potassium channels and causes hyperpolarization of the cell. In order for DSI to occur, the pre-synaptic cell has to be an inhibitory neuron. So, when the cell is hyperpolarized by the binding of endocannabinoids to the receptor, it suppresses the inhibitory effect of the pre-synaptic cell. This is a lot of information with a lot of acronyms, so I know it is easy to get lost. To try to help you understand DIS, I have included an image below that hopefully clears up any confusion.
Fig. 1. An image that shows the different steps of the endocannabinoid signaling cascade, including a description of what is happening. Drawn by H. Almlie.
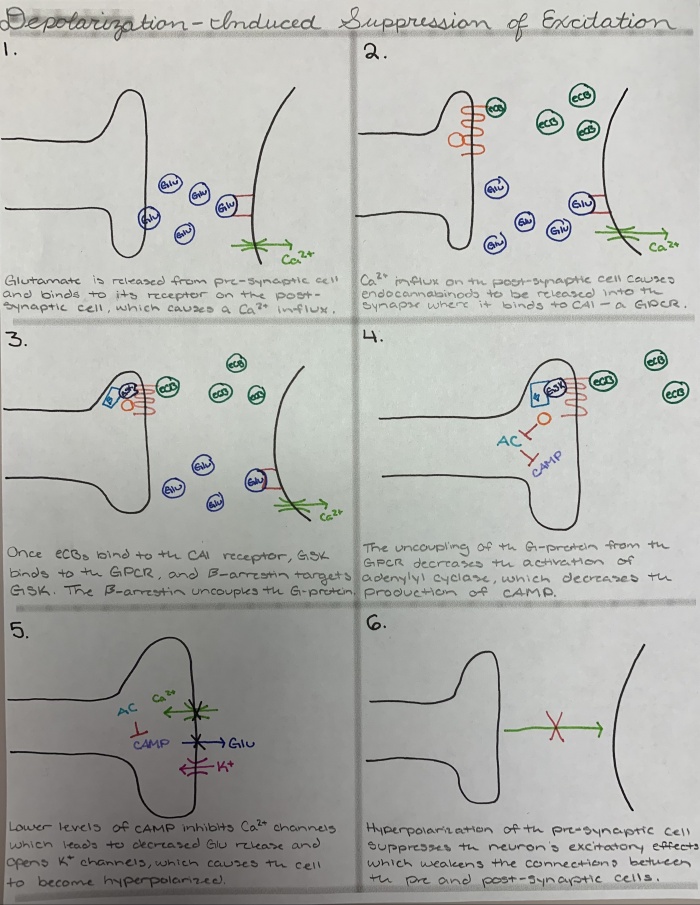
Depolarization-Induced Suppression of Excitation
DSE is not much different from DSI. The exact same signal cascade occurs both DSE and DSI, so I will not go through the entire signaling cascade in words. There is a difference in which ion enters the post-synaptic cell, but this is a minor difference that doesn’t affect the rest of the cascade. The only major difference is the result of the hyperpolarization. In the DSI signaling cascade, hyperpolarization of the pre-synaptic neuron suppresses its inhibitory effects. In DSE, the endocannabinoids are acting on an excitatory pre-synaptic cell. So, when the cell becomes hyperpolarized, the excitatory effects of the pre-synaptic neuron are suppressed. A summary image of the signaling cascade is shown below.
Fig. 2. An image of the various steps of the endocannabinoid signaling cascade, including descriptions of what is happening. Drawn by H. Almlie.
Synaptic Plasticity:
I have discussed the signaling pathway of the endocannabinoid system so that it is easier to visualize how synaptic plasticity is modulated by the system. So, I described in detail how depolarization-induced suppression of inhibition occurs, which we can then transfer to depolarization-induced suppression of excitation. Through these two mechanisms, the endocannabinoid system modulates synaptic plasticity. DSI helps to strengthen neural pathways decreasing the amount of inhibition that pathway experiences. DSE helps to weaken neural pathways by decreasing the amount of excitation experienced by neurons in that pathway. So, DSE and DSI help to modulate synaptic plasticity by essentially doing the opposite of what those neurons usually do. In this way, endocannabinoids play an essential role in modulating synaptic plasticity within the central nervous system.