
Cover Image Artstract designed by Cayley Borrud with the use of ChatGPT
Adolescence is often a rocky time full of firsts and figuring out identity. But what if in that volatile time, a teen decides to try vaping? It’s wrapped in colorful packaging, advertised to youth and the most used Tabacco product in youths [4]. How bad could just trying it be?
Image sourced from Alamy

The Neuroscience of Addiction
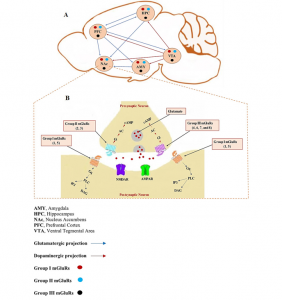
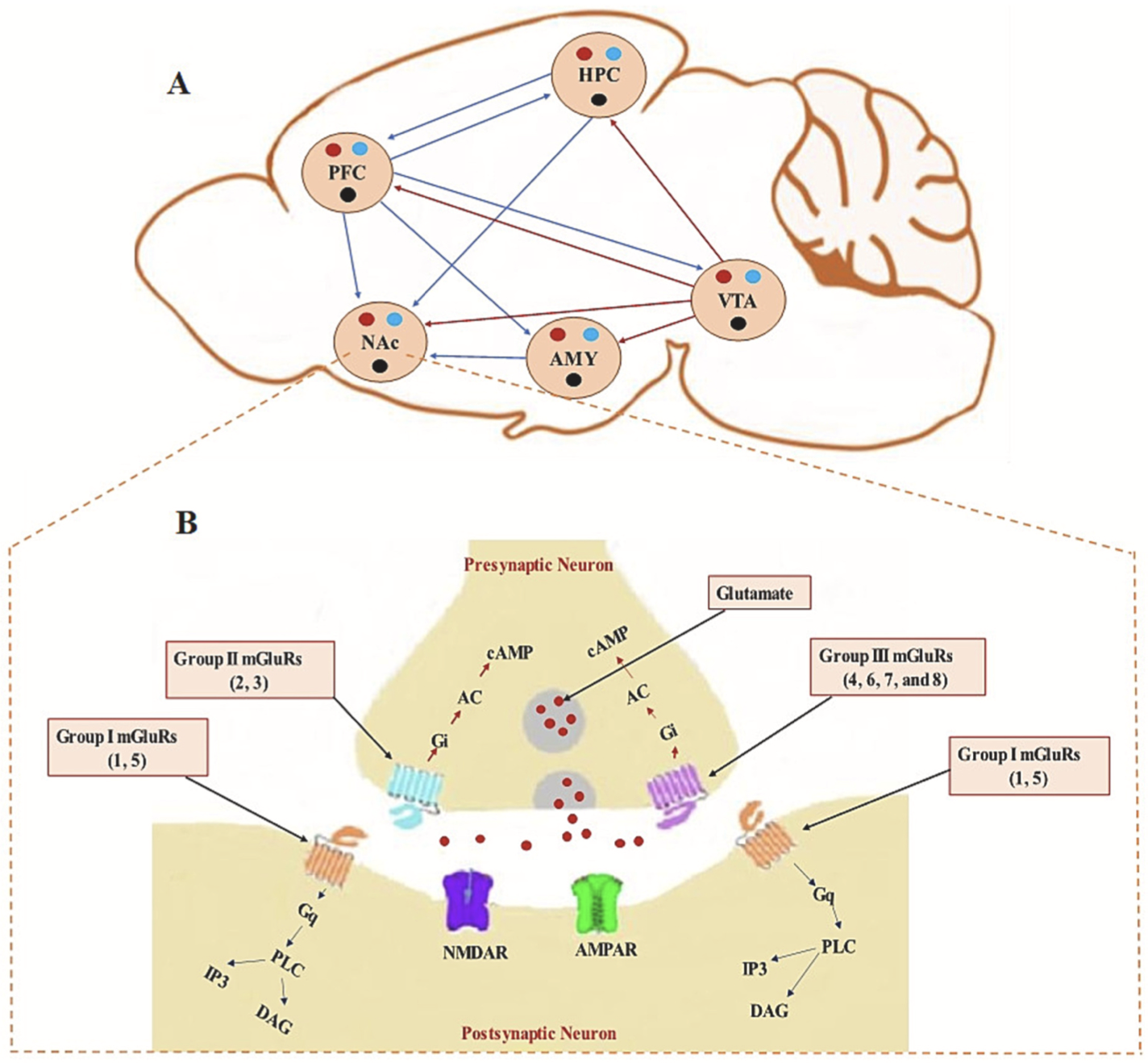
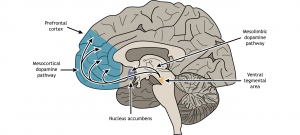
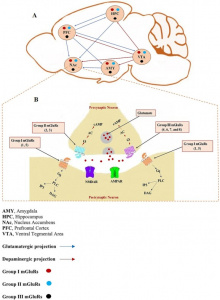
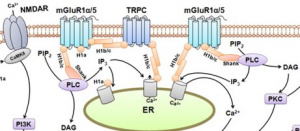
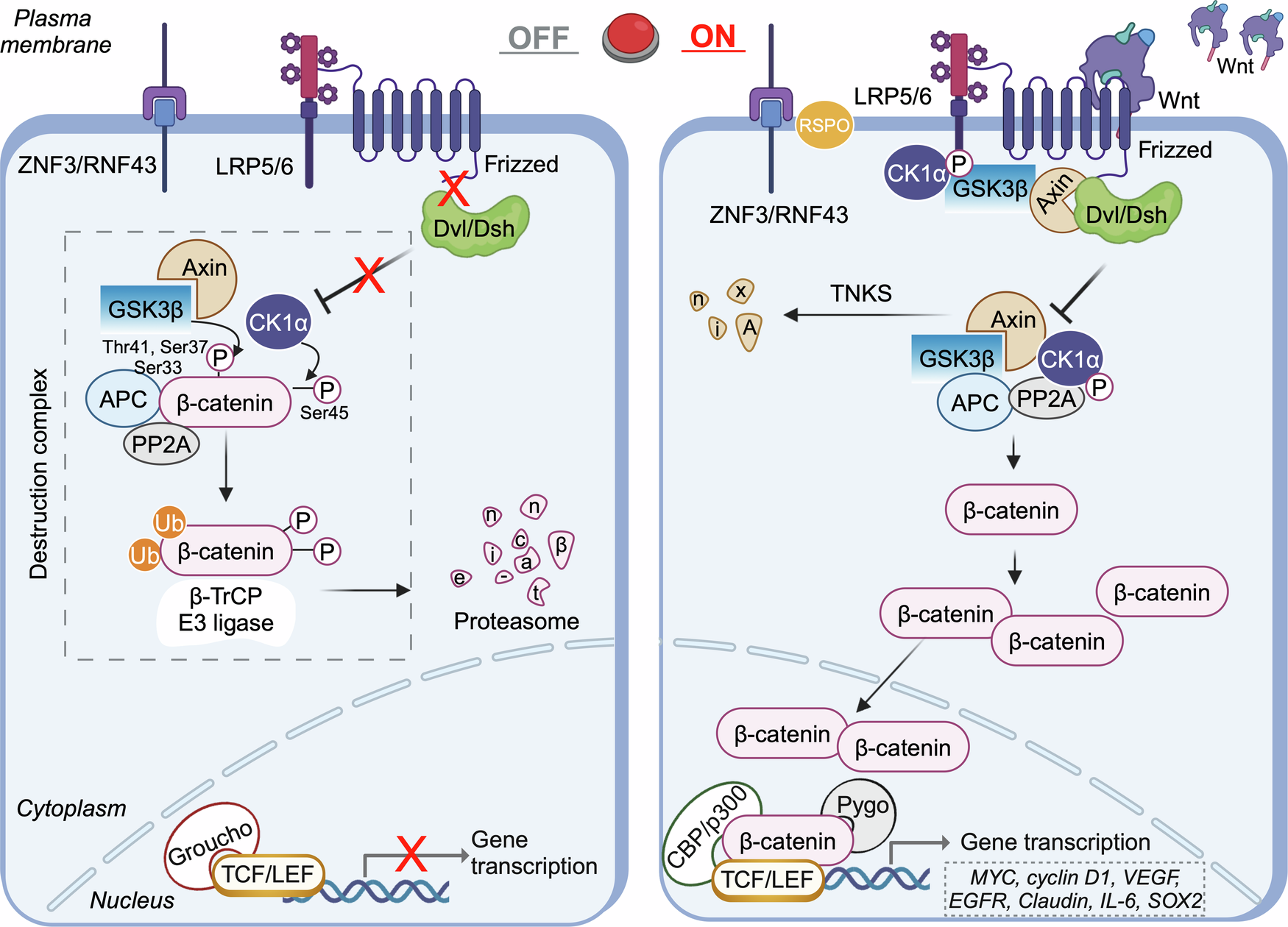
E-cigarettes are designed to make the person crave more. It alters the brains reward center through the disruption of metabotropic glutamate receptors (mGluRs). The paper by Mozafari et. al explains how mGluRs are very involved in addiction. The absence of this vital receptor leads to increased glutamate. Glutamate is a neurotransmitter that drives the drug seeking behavior. It strengthens the circuits in the brain which would normally be a good thing however, the circuits that it is strengthening in this case would be the memories of the substance [1].
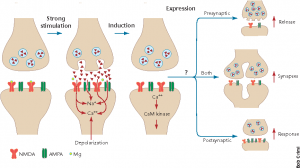
The brain could have structural changes such as increased dendritic spine density in the prefrontal cortex that can contribute to the formation of the ‘memory’ of vaping [3]. This is particularly a problem because it can make adolescents more likely to develop more addictions due to that rewiring of the brain. It also makes it much harder to stop their addiction since a non-fully developed brain is more likely to develop permanent addictions that follow them well into their adulthood [5].
Image sourced from Baker Institute
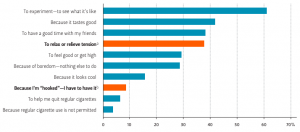
What started as curiosity driven experimentation would now be an addiction. This addiction can happen faster in teens than adults and it can greatly affect attention, impulse control and anxiety. The brain chemistry of the developing brain can change so rapidly that before nicotine is even consumed daily, withdrawal symptoms can appear [4].
The brain circuits involved in the development of addictions are constantly changing during adolescence which can lead to more risky behaviors [5]. What started as experimenting with e-cigarettes can quickly turn into experimentation and abuse of drugs and alcohol.
In adolescence, the dopamine signaling pathway is particularly sensitive and this pathway doesn’t reach full maturity until after adolescence [2]. This means that when a substance such as nicotine is taken, there would be a stronger reinforcement and quicker learned addictive behaviors [5]. The teenage brain is creating associations that vaping means reward and that association is the memory that’s hard to forget.
Now, let’s say, that teenager was able to stop vaping with the help of therapy and family support. That addictive pathway is still there and ready to be activated in moments of vulnerability or even from environmental cues associated with that nicotine memory [4]. Their brain is permanently changed. Unfortunately, adolescents don’t generally know that their actions are permantely changing their brain.
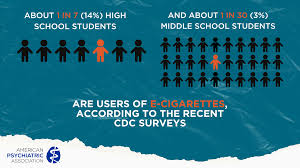
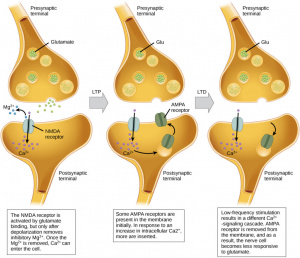
Image sourced from The American Psychiatric Association
Teens are biologically primed to become addicted therefore there needs to be more awareness and actions taken to protect them. There needs to be an urgency to protect teens before experimentation turns into long term consequences. By prioritizing prevention, regulation, and education, we can help protect developing brains during one of the most vulnerable stages of life.
Sources:
- Mozafari, R., Karimi-Haghighi, S., Fattahi, M., Kalivas, P., & Haghparast, A. (2023). A review on the role of metabotropic glutamate receptors in neuroplasticity following psychostimulant use disorder.Progress in Neuro-Psychopharmacology & Biological Psychiatry, 124, 110735. https://doi.org/10.1016/j.pnpbp.2023.110735
- Suri, D., Zanni, G., Mahadevia, D., Chuhma, N., Saha, R., Spivack, S., Pini, N., Stevens, G. S., Ziolkowski-Blake, A., Simpson, E. H., Balsam, P., Rayport, S., & Ansorge, M. S. (2023). Dopamine transporter blockade during adolescence increases adult dopamine function, impulsivity, and aggression.Molecular Psychiatry, 28(8), 3512–3523. https://doi.org/10.1038/s41380-023-02194-w
- Brown, R. W., & Kolb, B. (2001). Nicotine sensitization increases dendritic length and spine density in the nucleus accumbens and cingulate cortex.Brain Research, 899(1–2), 94–100. https://doi.org/10.1016/s0006-8993(01)02201-6
- (2024, October 17).E-cigarette use among youth. Smoking and Tobacco Use. https://www.cdc.gov/tobacco/e-cigarettes/youth.html
- Adolescents are neurologically more vulnerable to addictions | yale news. (2003, June 18). https://news.yale.edu/2003/06/18/adolescents-are-neurologically-more-vulnerable-addictions