Overview of the ALS
Amyotrophic Lateral Sclerosis or ALS is a fatal disease that affects motor neurons. It is defined by the degeneration of upper and lower motor neurons, but the severity, symptoms, age of onset and cognitive deficits vary from case to case. Ultimately, respiratory failure due to the degeneration of the motor neurons is what causes the death of ALS patients.
What causes the development of ALS?
ALS is classified into two different sub-types: familial ALS (fALS) and sporadic ALS (sALS). The familial forms of ALS account for 10% of total ALS diagnoses and stems from inheritable, autosomal genetic mutations. It is usually characterized by having a family member with ALS as well. Sporadic ALS cases make up the remaining 90% of ALS diagnoses. These patients have no family history of ALS and there is no direct inheritance of a genetic mutation. The average age of onset for sporadic ALS is 56 which is about 10 years later than for familial ALS which is 45.7. These two sub-types of ALS are clinically the same but differ in how the genetic mutations develop. There are genetic mutations found in fALS that are also found in sALS which cause the motor neuron degeneration. However, the difference is the family history of the patient. 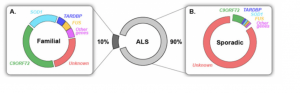
For example, a mutation of the SOD1 gene is found in both fALS and sALS. SOD1 is an antioxidant enzyme that helps defend against Reactive Oxygen Species (ROS) that the cell produces. When there is a mutation in this enzyme, it leads to the oxidative stress that is found in the pathology of ALS. Oxidative stress also can cause the unfolded protein aggregation that are found in the motor neurons of ALS patients.
The development of this disease can happen two different ways that are linked by common factors. Either oxidative stress and mitochondrial dysfunction cause changes in RNA metabolism leading to the motor neuron degeneration, or mutations in the proteins that bind RNA cause oxidative stress and mitochondrial dysfunction which also leads to motor neuron degeneration. The common links of these two different ways ALS can develop are the oxidative stress, RNA metabolism alteration and mitochondrial dysfunction. These ultimately cause the degeneration of the motor neurons by either protein aggregation, energy deficit or RNA dysfunction.
Caring for those with ALS
There are currently no treatments for ALS that can reverse the neurological damage that it causes. However, current medications aim at slowing the damage the disease causes and managing the pain the patient experiences. Breathing care is used to help patients breathe as their muscles become weaker. Physical therapy is also used to help with pain, walking and mobility to help the patient remain independent for as long as possible. Low-impact exercise is also used to maintain fitness and physical abilities. Other therapies used to manage the disease include occupational and speech therapy, nutritional support and psychological support.
In the late stage of ALS, most voluntary muscles are paralyzed (including those for breathing and swallowing). Patients in this stage usually require a ventilator for breathing and feeding tubes. The ALS association suggests hospice care for patients in the late stage of the disease. When patients received hospice care they were more likely to die in their preferred location (usually outside the hospital) and receive morphine. The final stage of ALS is the most physically and mentally distressing for the patient so it is important that their comfort level and desires are recognized.