
Comorbidities
Schizophrenia almost never arrives alone. In clinical practice, psychiatric comorbidities are usually the rule rather than the exception, and their prevalence is striking. Roughly 50% of patients experience significant depression at some point across the illness course, with PTSD and OCD corresponding to an estimated 29% and 23% prevalence among patients respectively. Furthermore, Substance use disorders carry a lifetime prevalence of approximately 47%, making them the most common comorbidity of all.1
That substance use figure deserves particular attention, as several competing explanations exist. Such explanations include mere chance, the use of substances as self-medication, substances directly precipitating psychosis, and shared neurobiological vulnerability. The self-medication hypothesis is intuitive but loses force in the era of better-tolerated second-generation antipsychotics as you would expect a decline in substance abuse rates – a decline which doesn’t exist. Interestingly, a meta-analysis suggested adolescent cannabis use as being linked to schizophrenia developing later in life.2 Nevertheless, any concrete link remains unsubstantiated. A few things cut across all of these comorbidities: each is associated with worse outcomes than schizophrenia alone, all appear across the full illness course, the neurobiological basis of each remains understudied, and treatment is largely trial and error without clear evidence-based guidance tailored to these subgroups.
Recent Work
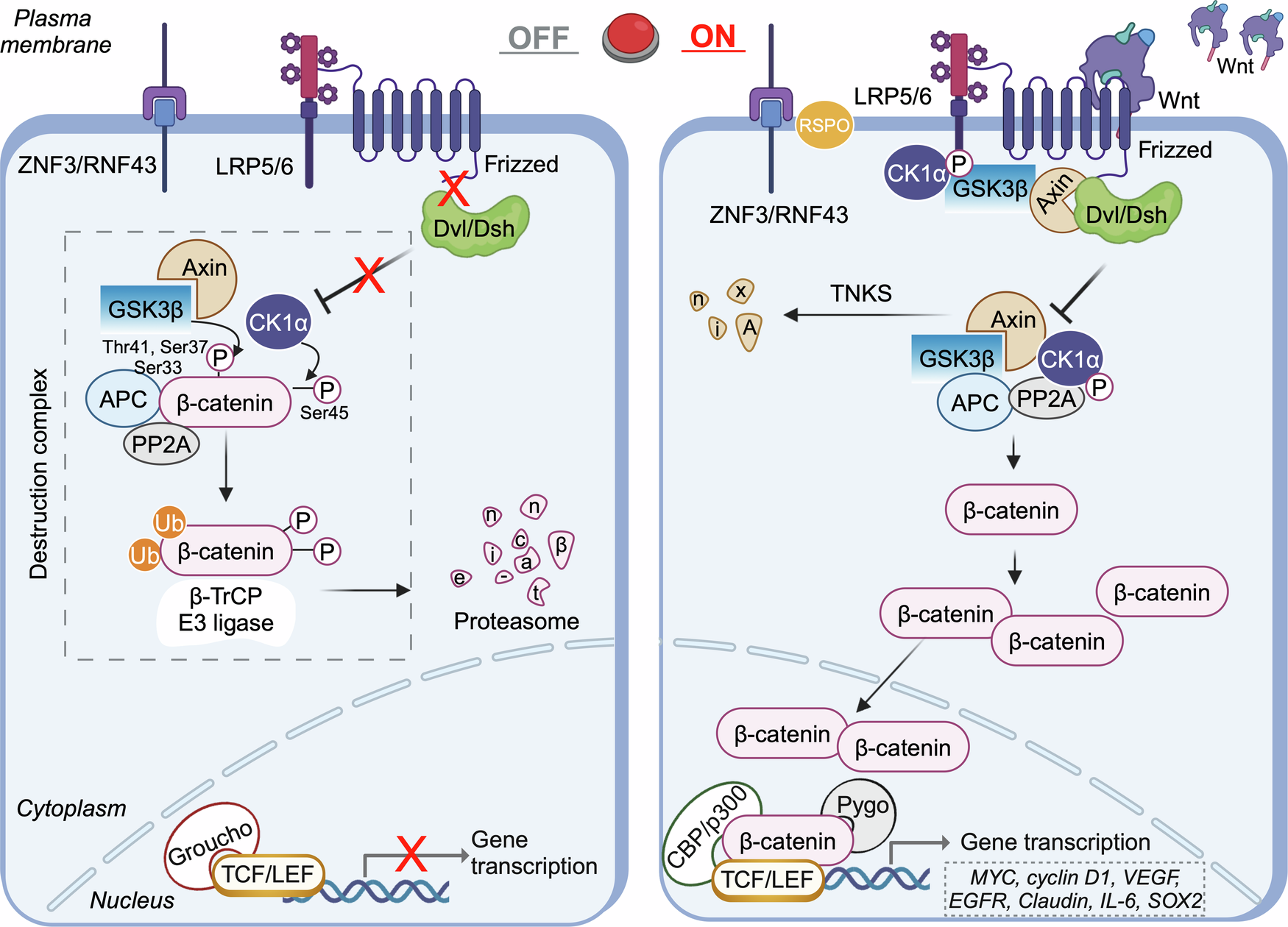
This is where emerging molecular research becomes relevant not as a solution, but as a framework for asking better questions. A 2013 review by Karun Singh in Clinical Genetics synthesized converging evidence implicating Wnt and glycogen synthase kinase 3 (GSK3) signaling in the biological basis of schizophrenia.3 The canonical Wnt pathway regulates β-catenin stability and downstream gene transcription, and plays a prominent role in neural development. The pathway depends on the stability of β-catenin: whether it gets broken down or or survives long enough to enter the nucleus. GSK3β is the key enzyme that drives that breakdown, so when Wnt signaling is active, or when GSK3β is blocked, β-catenin accumulates and gene transcription follows. This is illustrated clearly in Figure 1, where Wnt signaling being turned off results in the destruction of β-catenin, and Wnt being turned on results in the surplus of β-catenin which enters the nucleus.4 When disrupted, the consequences affect the very systems implicated in psychiatric vulnerability.
The pharmacological evidence, as laid out by Singh, is striking. Lithium, one of psychiatry’s oldest empirically validated treatments, works partly by directly inhibiting GSK3β and stabilizing β-catenin, effectively activating canonical Wnt signaling.5 Additionally, antipsychotics, whose primary target is the dopamine D2 receptor, modulate GSK3β activity through the Akt pathway as a downstream effect. Even metabotropic glutamate receptor agonists, a non-dopaminergic avenue, regulate GSK3 and accumulate Wnt pathway proteins with repeated treatment. The fact that these three mechanistically distinct drug classes converge on the same signaling network is not coincidental.
Human genetic findings reinforce this further. When scientists look at the genes associated with schizophrenia, they keep finding the same pattern. A gene called DISC1, one of the most well-known schizophrenia risk genes, works by blocking the same protein that lithium blocks, suggesting that what lithium does as a drug, this gene does naturally, and when it goes wrong, the consequences look like schizophrenia.6 Studies examining brain tissue from people who had schizophrenia have also found reduced levels of a protein called Akt1, and large-scale genetic studies keep flagging the same region.7 Taken together, these findings suggest that disruptions in this particular signaling network aren’t just a side observation; instead, they may be a genuine part of what actually causes schizophrenia in the first place.
What does this mean for the comorbidity problem?
The high rates of depression, anxiety, OCD, and substance use disorders seen alongside schizophrenia may not be coincidental — they may reflect a shared underlying disruption in Wnt/GSK3 signaling that plays out differently across neural systems. Because this pathway touches so many aspects of brain development and function, a single disruption early on could leave multiple systems vulnerable in ways that manifest as distinct but co-occurring disorders. This doesn’t solve the comorbidity problem, but it suggests these conditions may share a common biological origin rather than simply co-occurring by chance.