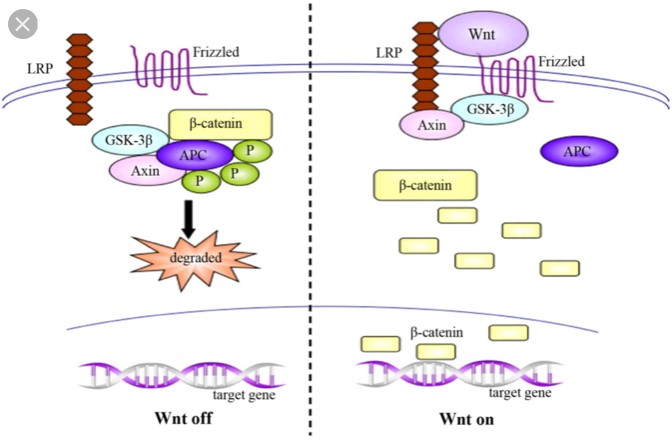
Schizophrenia is a devastating diagnosis, largely due to its lifelong nature and lack of effective treatment options. The underlying mechanism of schizophrenia is still largely unknown, but new evidence suggests a pathway that may be causing schizophrenia. While there is still much more to learn, this research has greatly increased the chance of new treatment options. This research implicated a pathway known as the canonical Wnt pathway. This pathway is present in nearly all organisms, indicating that it is highly important for proper development.
A large degree of the complexity associated with Schizophrenia is the fact that it is a developmental disorder, but symptoms don’t appear until adolescence. This progression also suggests that the disorder is somehow associated with the frontal lobe as this is the most active area of the brain during adolescent development. GS3KB is an important aspect of this pathway due to its role in regulation of gene transcription. The dysfunction associated with Schizophrenia occurs when GSK3B is overactive and cannot regulate transcription properly. The pathway on the left is more often occurring on the left than on the right in the brains of people with schizophrenia, leading to dysfunction and schizophrenia symptoms.
What is Schizophrenia?
Schizophrenia is characterized by a month or longer period of two or more of the following:
- Delusions
- Hallucinations
- Disorganized Speech
- Grossly Disorganized or catatonic behavior
- Negative Symptoms such as flat affect
People often think of Schizophrenia as having vivid hallucinations and delusions but don’t often realize there are negative symptoms as well. Negative symptoms include flat affect, lack of social interest, and little motivation. These symptoms are especially common after use of anti-psychotic medications, because the positive symptoms of the disorder have been treated.
Schizophrenia treatment
Anti-psychotics are a common treatment for Schizophrenia, but they are far from perfect. Common side effects of Schizophrenia include:
- Dry mouth
- Stiffness and shakiness
- Restlessness
- Weight gain
- Sleepiness and slowness
- Sexual dysfunction
- Constipation
- Blurry vision
- Uncomfortable jaw, lip, and tongue movements
In addition, the nature of the disease makes people less likely to adhere to their medication schedule. People start to feel better and don’t realize how much of an impact their medications make. Delusions may also convince people not to take their medications. New forms of anti-psychotics can be administered as injections, making adherence to medication less of a challenge.
Are people with Schizophrenia dangerous?
People who have not been exposed to mental illness and who lack education often assume that people with certain mental illnesses are more dangerous than the general public. This could not be further from the truth. People with mental illness are actually more likely to be victims of violence than people without. Schizophrenia is widely understood, but more education about it could lead to a collective understanding that people with Schizophrenia are still people who should be treated with respect, dignity, and compassion.