Many of us out there are coffee drinkers. I myself have at least one cup every morning. I will admit, yes, I am slightly addicted to my morning coffee. But am it’s not really the coffee I am addicted to. It’s the caffeine in the coffee that wakes me up every morning.
According to the American Psychiatric Association, addiction is a disease that is caused by compulsive substance use despite harmful consequences. People who have an addiction or multiple addictions, are no longer able to control their will to use the substance or not to. The most common substance people become addicted to are drugs including alcohol, cocaine, methamphetamine, and opioids, among several others.
People use drugs to feel good, feel better, to do better, or just out of sheer curiosity. Some people just want to feel good or to experience the high that accompanies several drugs. Others, such as those with anxiety or depression, use drugs because it makes them feel better. Some use because they believe it helps them perform better at certain tasks. But no matter the reason people use drugs, the same pathways are activated in the brain.
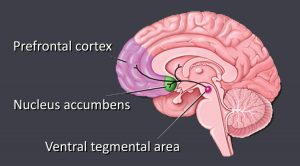
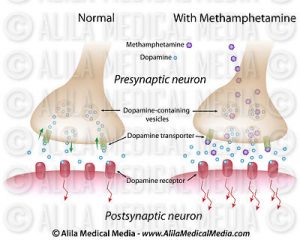
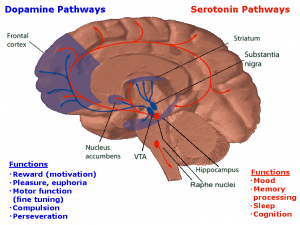
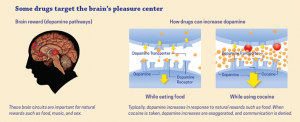
The pathway that gets activated when a person uses a drug is the mesolimbic dopamine pathway. This is also known as the reward pathway. This pathway involves the release of dopamine. Rewards such as food, sex, or some sort of drug increase the concentration of dopamine in the brain.

When someone drinks a cup of coffee, this same mesolimbic pathway releases dopamine. This causes the person to feel good. Because of this, the person associates drinking coffee with feeling good. The more coffee the person drinks, the more caffeine they consume. Caffeine is a stimulant that causes chemical changes in the brain. Consuming caffeine on a daily basis can build up a tolerance to the caffeine, requiring more and more to feel the same effects as before. When someone like this does consume caffeine one day, they may experience a headache or shakiness, because the body is not used to not having caffeine in it.
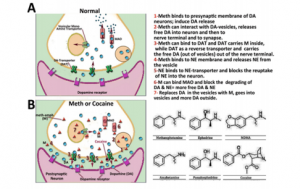
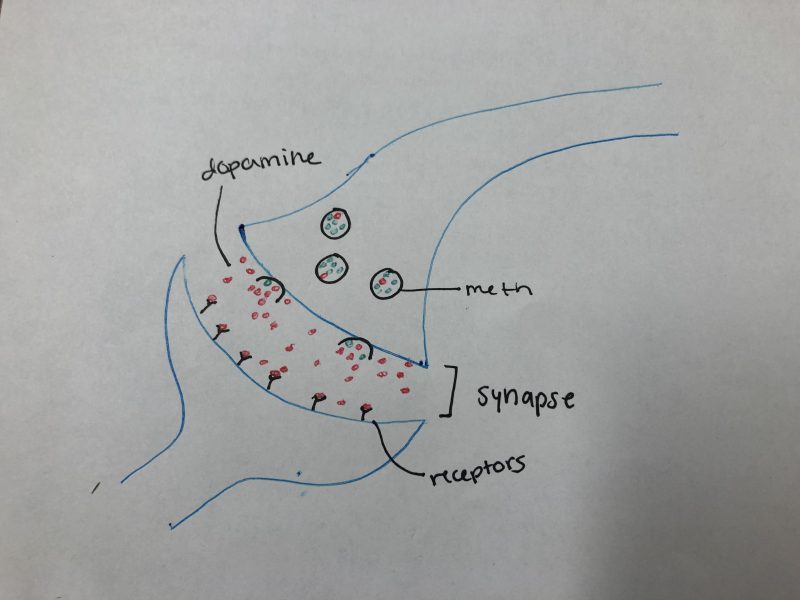
So, what about drugs such as cocaine? Cocaine is a powerful stimulant, just like caffeine. It acts as a mood modulator and anti-depressant. So far this sounds pretty good. But the effects of cocaine only last for about 30 minutes. After that, to feel the same effects, more of the drug would have to be consumed. When cocaine is consumed, it binds to dopamine transporters on the cell’s membrane. Here, it blocks the cell from from being able to reuptake the dopamine molecules that are present from being released from the mesolimbic pathway. This further increases the concentration of dopamine and its probability of binding to its receptors. So, similarly to caffeine, a person has to take more and more to feel the same effects as the first time.
In the end, caffeine addictions and cocaine addictions arise from the same cause: dopamine. They both call for the need to use more and more. They both can lead to withdrawal symptoms when not consumed. The difference between the two is that one is more socially acceptable than the other. More people consume caffeine every day than cocaine. So how come one is seen as a problem and the other is not?
https://www.psychiatry.org/patients-families/addiction/what-is-addiction
https://neuroscience.mssm.edu/nestler/brainRewardpathways.html
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1920543/
https://www.addictioncenter.com/stimulants/caffeine/
Images sourced from:
<p style=”font-size: 0.9rem;font-style: italic;”><img style=”display: block;” src=”https://mir-s3-cdn-cf.behance.net/project_modules/1400/4138b970842723.5bb14a8d0d607.jpg” alt=”Caffeine logo for a cafe”><a href=”https://www.behance.net/gallery/70842723/Caffeine-logo-for-a-cafe”>”Caffeine logo for a cafe”</a><span> by <span>Hassan Nasser</span></span> is licensed under <a href=”https://creativecommons.org/licenses/by-nc-nd/4.0/?ref=ccsearch&atype=html” style=”margin-right: 5px;”>CC BY-NC-ND 4.0</a><a href=”https://creativecommons.org/licenses/by-nc-nd/4.0/?ref=ccsearch&atype=html” target=”_blank” rel=”noopener noreferrer” style=”display: inline-block;white-space: none;margin-top: 2px;margin-left: 3px;height: 22px !important;”><img style=”height: inherit;margin-right: 3px;display: inline-block;” src=”https://search.creativecommons.org/static/img/cc_icon.svg” /><img style=”height: inherit;margin-right: 3px;display: inline-block;” src=”https://search.creativecommons.org/static/img/cc-by_icon.svg” /><img style=”height: inherit;margin-right: 3px;display: inline-block;” src=”https://search.creativecommons.org/static/img/cc-nc_icon.svg” /><img style=”height: inherit;margin-right: 3px;display: inline-block;” src=”https://search.creativecommons.org/static/img/cc-nd_icon.svg” /></a></p>
https://www.google.com/url?sa=i&source=images&cd=&ved=2ahUKEwiC7I3v9NPlAhUBfFAKHSTyBCQQjRx6BAgBEAQ&url=https%3A%2F%2Fgeneticliteracyproject.org%2F2017%2F11%2F30%2Fbrain-addiction-why-stopping-drug-use-difficult%2F&psig=AOvVaw3sOf2ffVhv_1qJFFUvcFQ6&ust=1573072194903649

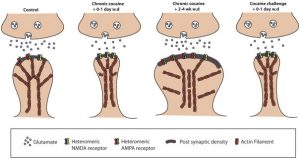
 factor causes more proteins to be produced than normal and affects the neural connections within the brain. Now, understanding slightly better what happens deep in the brain. So, is addiction within a person’s control?
factor causes more proteins to be produced than normal and affects the neural connections within the brain. Now, understanding slightly better what happens deep in the brain. So, is addiction within a person’s control?


 [5]
[5]