Abstract created by Gabe Sparks
Obesity is often labeled as a simple imbalance between the calories we consume and the calories we burn, but this disease runs much deeper than public perception suggests. It’s not just about food intake; it’s also about complex processes happening within the brain. Recent research shows that inflammation in certain brain regions can disrupt normal signaling in neural pathways, contributing to the development of obesity.1
One of the most affected areas is the hypothalamus, a region that plays a critical role in regulating feeding behavior and energy expenditure.1 For our bodies to maintain a healthy balance between the energy we take in through food and the energy we burn through physical activity the hypothalamus needs to function properly. When this region becomes inflamed, the signaling from key hormones like insulin and leptin can become impaired. These disruptions confuse the body’s internal cues for hunger and energy use, setting the stage for weight gain and metabolic dysfunction.
This is just a brief look at how obesity is far more complex than many realize. The story goes beyond diet and exercise. It reaches into the brain itself, revealing layers of complexity we’re only beginning to understand.
Understanding the Science
To understand the background of this metabolic disease, there are two key scientific areas to focus on: the specific neurons responsible for regulating energy balance, and the pathways disrupted by inflammation that lead to dysfunction in this system.
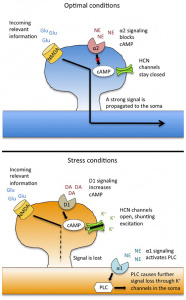
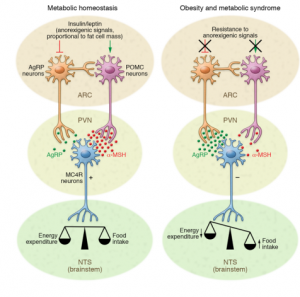
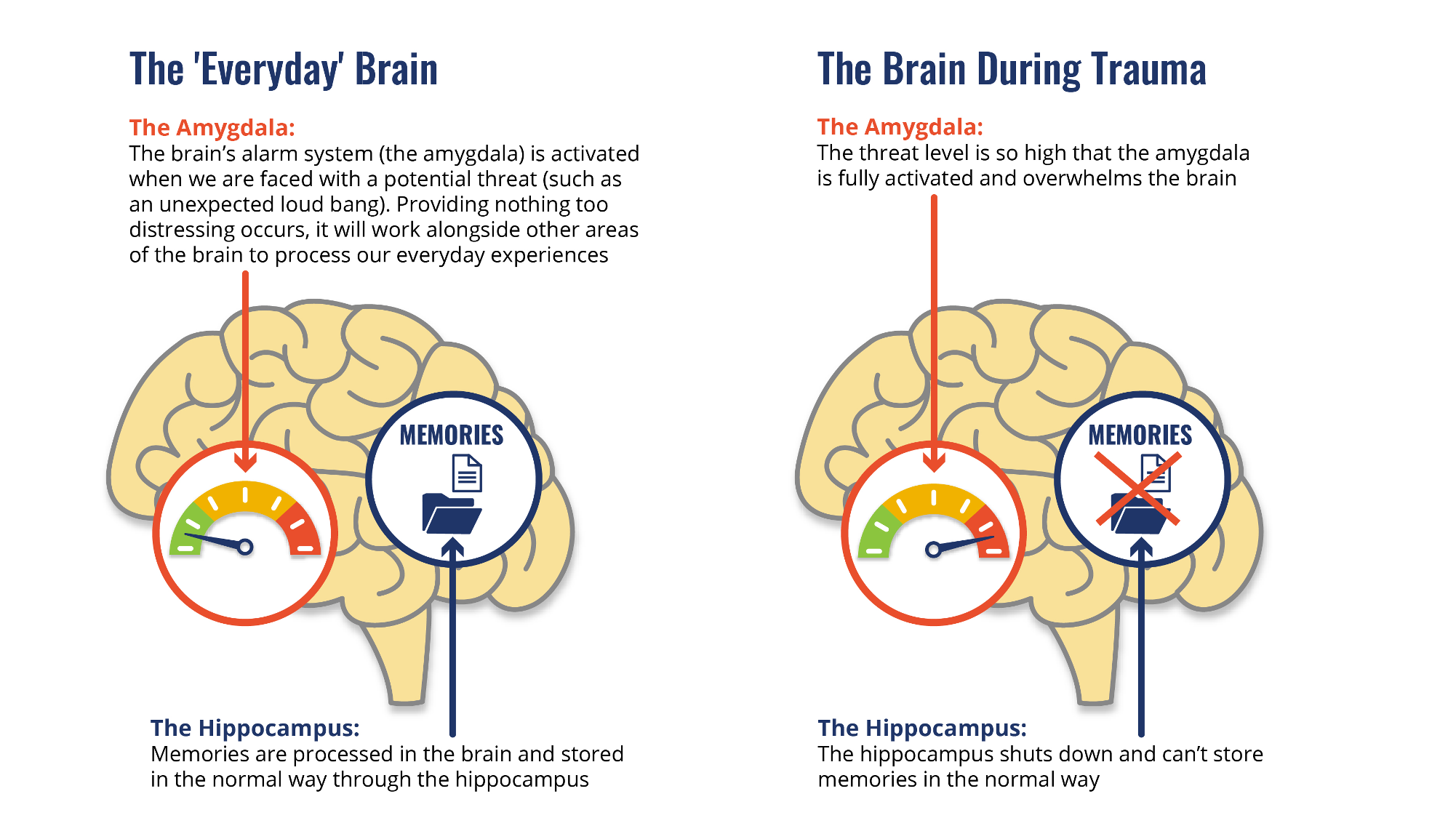
The two main types of neurons involved in energy homeostasis are AgRP (Agouti-related peptide) and POMC (pro-opiomelanocortin) neurons.1 Both are located in the arcuate nucleus (ARC) of the hypothalamus and play crucial, but opposite, roles in managing food intake and energy expenditure. When functioning properly, as shown on the left side of Figure 1, these neurons maintain a healthy balance between energy coming in and energy being used.

Figure 1.Diagram illustrating the neurons involved in metabolic homeostasis and the disruptions seen in obesity and metabolic syndrome.
However, in obesity and metabolic syndrome, this balance is disrupted. Overactivation of AgRP neurons results in increased signaling to the paraventricular nucleus (PVN), leading to reduced energy expenditure and increased food intake.1 This dysregulation is one of the earliest signs of hypothalamic inflammation and is a driving force in the development of metabolic disease.
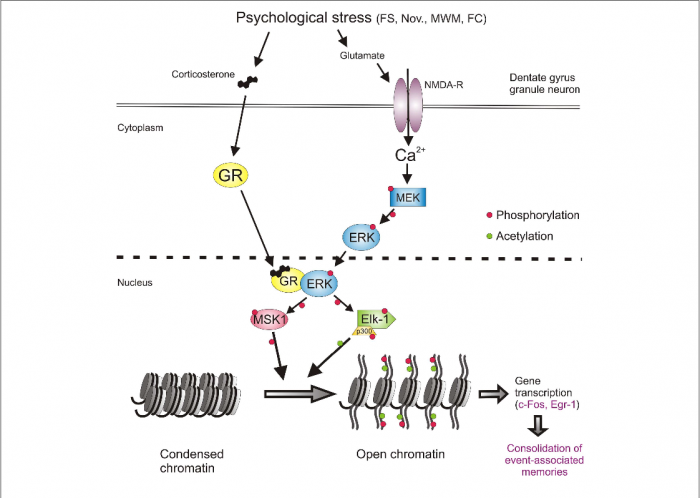
A deeper look into the molecular level reveals how this disruption happens. In a healthy brain, hormones like insulin and leptin bind to their receptors on hypothalamic neurons to help regulate appetite and metabolism. Insulin works by suppressing the activity of a protein called FOXO1, which promotes POMC expression (appetite-suppressing) and reduces AgRP/NPY expression (appetite-stimulating). Leptin activates STAT3, which similarly boosts POMC while suppressing AgRP/NPY activity.1
But in states of chronic inflammation, often triggered by molecules like TNF-α or saturated fats, these hormonal pathways are disrupted. TNF-α activates proteins such as JNK and NF-κB, which interfere with normal insulin signaling.1 This reduces the brain’s sensitivity to both insulin and leptin, weakening their ability to regulate hunger and metabolism. Additionally, inflammation increases the expression of SOCS3, a molecule that further blocks insulin and leptin signaling, reinforcing resistance. The result is a system that no longer responds appropriately to signals of satiety and energy balance, leading to overeating and metabolic dysfunction. These different pathways can be found in Figure 2.1

Figure 2. Diagram depicting the molecular pathways of metabolic inflammation in the hypothalamus.
The Start of Inflammation: Diet, Stress, and Modern Life
Inflammation is typically seen as a natural response to injury or infection, but chronic inflammation, especially in the brain, can be harmful. Today’s modern lifestyle, particularly our diet and stress levels, often acts as a continuous trigger, keeping our immune system in a low-grade, overactive state.2 This persistent inflammation contributes to the disruptions in neural pathways that lead to metabolic diseases.
The American environment plays a significant role in the rise of these conditions. Over time, we’ve witnessed a dramatic shift in the standard diet, marked by excessive consumption of saturated fats, refined carbohydrates, and trans fats, while nutrient-dense foods have become less common. The decline in dietary quality isn’t the only factor at play, though. The increasing presence of stress in our daily lives is another major contributor. The modern world constantly activates the HPA axis, releasing stress hormones like norepinephrine and cortisol.2 Stressors such as academic pressures, financial anxiety, or information overload have led to the prolonged production of pro-inflammatory cytokines, while simultaneously reducing the body’s ability to properly shut off the inflammatory response.
As a result, the combination of poor dietary habits and chronic stress has created an environment that fosters persistent inflammation, which ultimately disrupts the brain’s ability to regulate metabolism and appetite effectively.2
Here is a link to an article that further explores chronic inflammatory diseases and our current lifestyle.
Conclusion: Rethinking Obesity and Metabolic Health
The rise in obesity and metabolic diseases is far more complex than the public’s general understanding. As we’ve discussed, hypothalamic inflammation plays a pivotal role in the disruption of critical neurological pathways that regulate both hunger and energy expenditure. The modern American lifestyle provides an ideal environment for this inflammation, contributing to the growing prevalence of metabolic disorders.
By diving deeper into the mechanisms behind these diseases, we can shift public perception and broaden our approach to addressing them. Tackling the root causes of brain inflammation such as improving nutrition, managing stress, and making healthier lifestyle choices is crucial for long-term change. While the path forward may require significant effort, it’s a challenge we, as a society, must take on to build a healthier future for all.
References
(1) Jais, A.; Brüning, J. C. Hypothalamic Inflammation in Obesity and Metabolic Disease. Journal of Clinical Investigation 2017, 127 (1), 24–32. https://doi.org/10.1172/JCI88878.
(2) Bosma-Den Boer, M. M.; Van Wetten, M. L.; Pruimboom, L. Chronic Inflammatory Diseases Are Stimulated by Current Lifestyle: How Diet, Stress Levels and Medication Prevent Our Body from Recovering. Nutrition and Metabolism. 2012. https://doi.org/10.1186/1743-7075-9-32.














:max_bytes(150000):strip_icc()/how-to-reduce-stress-5207327_FINAL-907db114a640431ba1e8ecbb9e81b77f.jpg)